ANTIVIRAL AGENTS pharm
advertisement

ANTIVIRAL AGENTS
Viruses consist of either ds or ss DNA RNA enclosed a protein coat called a capsid.
Some viruses also possess a lipoprotein envelope.
Viruses are obligate intracellular parasites; their replication depends on primarily on systemic processes of the host cell. most anti-viral agents have
proved of little use therapeutically since the virus uses host-cell metabolic reactions and thus, for the most part, anti-viral agents will also be anti-cell agents. Thus,
the alternative approach of stimulating the host's immune responses using vaccines has been most often pursued. Nevertheless, there are activities (i.e. enzymes)
that are virus-encoded and therefore offer potential virus-specific targets. This is particularly the case with the viruses that have large genomes and code for their own
replication enzymes. Even so, unfortunately, many anti-virals that are apparently effective in vitro are ineffective in vivo.
A successful anti-viral drug should:
(i) interfere with a virus-specific function (either because the function is unique to the virus or the similar host function is much less susceptible to the drug) or
(ii) interfere with a cellular function so that the virus cannot replicate. To be specific, the anti-viral drug must only kill virus-infected cells. This could be done by
restricting drug activation to virus-infected cells.
An ideal drug should be: Water-soluble
Stable in the blood stream
Easily taken up by cells
An ideal drug should NOT be:
Toxic
Carcinogenic
Allergenic
Mutagenic
Teratogenic
Toxicity of an anti-viral drug may be acceptable if there is no alternative: such as, for example, in symptomatic rabies or hemorrhagic fever
Obviously, a good drug must show much more toxicity to the virus than the host cell. We measure selectivity by the therapeutic index of the drug
Therapeutic index (T.I.): Minimum dose that is toxic to cell
Minimum dose that is toxic to virus
Effective drugs have a T.I. of 100-1000 or better
Consequently,to be effective, antiviral agents must either block viral entry into or exit from cell or be active inside host cell.
Viral replication consist of many steps:
1. ardsorption to and penetration into susceptible host cells
2. uncoating of viral nucleic acid
3. sythesis of early regulartory proteins, eg eg, nucleic acid polymerases
4. sythesis of RNA or DNA
5. sythesis of late structural proteins
6. assembly (maturation) of viral particles
7. release from the cell.
NB
Antiviral drugs can target any of this steps.
Table showing the stages of viral replication and the possible targets of action of antiviral agents
STAGE OF REPLICATION
CLASSES OF SELECTIVE INHIBITORS
Cell entry
Attachment
Soluble receptor decoy, antireceptors
Penetration
Antibodies, fussion protein inhibitors
Uncoating(release of viral genome)
Ion channelblockers, capsid sterbilizers
Transcription of viral genome
Transcription of viral mRNA
Replication of vira genome
Translation of viral proteins
Inhibitors of DNA viral polymerase, RNA polymerase,
reverse transcriptase, helicase, primase, or intergrase.
Regulatory proteins
Inhibitors of regulartory proteins
Post translational modifications
Proteolytic cleavage
Protease inhibitors
Assembly of virion components
Interferons, assembly protein inhibitors
Interferons, antisence oligonucleotides, ribozymes
Release
Budding cell lysis
Neuraminidase inhibitors, antiviral antibodies,cytotoxic
lymphocytes
Nomenclature of currently approved antiviral agents (excluding antiviral agents)
GENERIC NAME
DOSAGE FORMS
ANTIHERPES VIRUS AGENTS
IV, O,T,Ophth
Acyclovir
IV,T
Cidofovir
T
Docosanol
O
Famciclovir
IV, O
Foscarnet
Intravitreal
Fomivirsen
IV, O, Intravitreal
Ganciclovir
Ophth
Idoxuridine
T, IV
Penciclovir
Ophth
Trifluridine
O
Valacyclovir
Ophth, IV
Vidarabin
ANTIINFLUENZA AGENTS
Amantadine
Oseltamivir
Rimantadine
zanamivir
OTHER ANTIVIRAL AGENTS
Fomivirsen
Imiquimod
Interferon – alfa(alfa – 2a, alfa – 2b, alfan3, alfacon – 1, alfa
n1)
Peginterferon alfa-2b
Peginterferon alfa – 2a
Lamivudine
Ribavirin
O
O
O
Inhaled
Intravitreal
T
C/IM, intralesional
SC
SC
O
O, Inhaled, IV
ANTIHERPS VIRUS AGENTS
Infection with herpe simplex virus type 1 typically causes disease of the mouth, face, skin, esophagus or brain while type 2 usually causes infections
of the genitals, rectum, skin,hands, or meninges.
In either case the infection may be primary or can result from activation of alatent infection.
Acyclovir
Cyclic guanine nucleoside analog that lacks 3’-hydroxyl on the side chain
Approved in 1982.
Acyclovir {9-[(2-hydroxyethoxy)-methyl]guanine} is an antiviral agent which has been shown to be effective in the prevention and treatment of
herpes simplex virus (HSV) and varicella-zoster virus (VZV) infections . It is also used for prophylaxis of cytomegalovirus infection following solid
organ transplantation. It is active against HSV1, HSV 2 and VSV. Invitro activity against EBV and CMV and HSV6 is present but comparatively
weak.
Acyclovir exhibits a selective inhibition of herpes virus replication and has potent clinical antiviral activity against HSV and VZV, which can cause
significant morbidity and mortality due to primary infection or to reactivation following long-term latency in pediatric oncology and pediatric bone
marrow/stem cell transplantation . Children with malignant diseases who experience prolonged periods of myelosuppression due to cytotoxic
chemotherapy or hematopoietic stem cell transplant are highly susceptible to invasive viral infections . The use of indwelling vascular catheters
further increases the risk of development of invasive viral infections among immunosuppressed children .
Formulations: capsules, as an ointment, and powder to be reconstituted for intravenous use. The efficacy of oral acyclovir may be limited by its low
oral bioavailability (F), which is between 15% and 30% . Valacyclovir (valaciclovir), the l-valyl ester prodrug of acyclovir, is rapidly and extensively
converted to acyclovir after oral administration and can increase the oral F value of acyclovir up to 50%.
Mechanism of action
Acyclovir inhibits viral DNA synthesis
Acyclovir requires three phosphorylationsteps for activation(it’s a pro drug)
Its converted is to monophosphate, by the virus specified thymidine kinase( the affinity of acyclovir for HSV thymidine kinase is 200-fold greater
than for the mammalian enzyme) and then to di- and triphosphate compounds by the hosts cellular enzymes.
Because it requires the viral kinase for the initial phosphorylation , acyclovir is selectively activated and the triphosphate accumulates only in
infected cells.
Acyclovir inhibits viral DNA synthesis by two mechanisms:
1. competitive inhibition deoxyguanosine triphosphate for the viral DNA polymerase with binding to DNA template as and irreversible complex.
2. chain termination following incorporation into the viral DNA.
Pharmacokinetics
Oral bioavailability of acyclovir ranges from 10% - 30% and decreases with increasing dose. Peak plasma concentrations average 0.4 to 0.8 µgm/l
after 200mg and 1.6µg/ml 800mg doses.
Following intravenous administration, peak plasma concentration average 9.8 µg/ml after 5mg/kg per 8 hrs and 20.7µg/ml after 10mg/kg per hrs.
Percutaneous absorption of acyclovir after topical administration is low.
Valacyclovir is converted rapidly and virtually completely to acyclovir following oral administration in healthy individuals. This conversion is
thought to be due to first pass intestinal and hepatic metabolism.the relative oral bioavailability of acyclovir increases 3 - 5 -fold to approximately
70% following valacyclovir administration.
Acyclovir distributes widely in the body fluids including vesicular fluids, aqueas humor and cerebrospinal fluids. Compared to plasma salivary
concentrations are low and vaginal secretion concentration vary widely. Acyclovir is contacted in brest milk, amniotic fluid, and placenta. New born
plasma levels are similar to maternal ones. Following administration of acyclovir or valacyclovir, approximately 10% of its dose is metabolized in
the liver. Acyclovir is predominately eliminated by renal excretion via glomerular filtration and tubular secretion. A patient's renal function
significantly influences the pharmacokinetics of acyclovir, with the mean systemic clearances (CLs) of acyclovir being 327 ± 80, 248 ± 62, and 190 ±
62 ml/min/1.73 m2 in adult patients, with estimated creatinine CLs (CLCRs) of >80, 50 to 80, and 15 to 50 ml/min/1.73 m2, respectively).
The mean plasma t ½ acyclovir is 2.5 hours in adults and 4 hours in neonates. T ½ increases to 20 hrs in anuric patients.
Renal excretion of un metabolized acyclovir by glomerular filtration and tubular secretion is the principal route of elimination. Less than 15% is
excreted as 9-crboxymethylguanine or minor metabolites. For all indications reduction in dosing is required for PTs with renal issuficince.
Therapeutic uses
Oral acyclovir is used in treatment of primary and recurrent genital herpes
200mg 5x daily of acyclovir shortens the duration of symptoms, the time of viral shedding and the time to resolution of lesions.by approximately 5
days in primary HSV infections. Clinical response is less dramatic in the treatment of recurrent disease with the shortening of the time cause by 12 days.
Treatment of primary genital herpes does not alter the frequency or severity of recurrent outbreaks.
Intravenous acyclovir is the treatment of choice for herpes simple encephalitis and neonatal hsv infection. Its also effective for treatment of
primary and recurrent HSV infections but is reserved fo patients with severe disease and those who cant swallow pills.
Topical acyclovir is much less effective than oral therapy for primary HSV infection. Its of no benefit in treating recurrences.
Because VZV is less susceptible to acyclovir than HSV, higher doses are required.
In immunocompromised patients with zooter IV acyclovir reduces the incidence of cutaneous and visceral dissemination.
Oral acyclovir(400mg every 12 hrs) is effective for chronic suppression of recurrent genital herpes in PT with frequent outbreaks. Recurrence of
outbreaks may resume upon discontinuation of suppressive acyclovir.
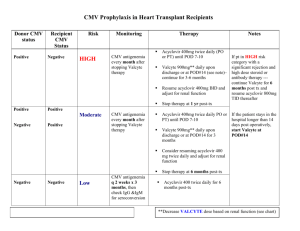
The benefit of acyclovir in prevention of CMV in transplant PTs is controversial.
Intravenous and high-dose oral acyclovir has been the gold standard for many children requiring treatment and/or prevention of herpes simplex virus
(HSV) and varicella-zoster virus (VZV) infections. However, intravenous acyclovir requires hospitalization, and oral acyclovir has limited
bioavailability, necessitating frequent dosing, and may not provide maximum therapeutic benefit especially in immunocompromised hosts requiring
higher plasma drug levels.
Unwantedeffects
Acyclovir generally is well tolerated. Topical acyclovir in a polyethylene glycol may cause mucosal irritation and transient burning when applied to
genital lesions.
Oral acyclovir has been associated with nausea, diarrhea rash or headache.
Intravenous infusion may be associated with renal insufficiency or neurologic toxicity(including tremors and delirium); however this are uncommon
with adequate hydration and avoidance of rapid infusion rate
Moreover, renal impairment is a commonly reported adverse effect of acyclovir. For intravenous dosing, acyclovir must be infused over 1 hour to
avoid high concentrations, as acyclovir sodium can precipitate as crystals in the renal tubules, with the risk of tubular damage. Hence, the dosage of
acyclovir or valacyclovir may need to be reduced, and the dosing interval may need to be increased, in patients with renal impairment to reduce the
risk of acyclovir-induced neurotoxicity due to drug accumulation.
Daily use of acyclovir for suppression of genital herpes for more than 10 yrs has not been associated with unwanted effects.
There has been no evidence of teratogenicity in pregnancy.
Acyclovir demonstrates high interindividual variability (IIV) in its treatment response, with a wide range of adverse reactions. While the current
dosage recommendations for acyclovir and valacyclovir are based on body weight (5 mg/kg and 10 mg/kg of body weight, respectively), there are no
specific recommendations for reduction of the acyclovir dose in pediatric patients with renal impairment, with the exception of some data for
neonates. Valacyclovir is used in the clinical setting because of its significantly high F value compared with that of oral acyclovir. However,
information regarding its optimal use in children is limited because pharmacokinetic data are lacking and the safety and effectiveness of valacyclovir
in children have not been established
Resistance
Acyclovir resistance in HSV has been linked to :
absence or partial production of viral thymidine kinase(most common),
altered thymidine kinase substrate specificity(e.g phosphorylation of thymidine but not acyclovir).
altered viral DNA polymerase
caused by point mutations or base insertion or deletions.
Famciclovir
Penciclovir, a nucleoside analogue, possesses potent in vitro antiviral activity against HSV types 1 and 2 and VZV . Famciclovir, the oral prodrug of
penciclovir, is currently approved for the treatment of herpes zoster and herpes labialis, treatment or suppression of genital herpes in
immunocompetent adult patients, and treatment of HSV infections and herpes zoster in immunocompromised adults.
Is the diacetyl ester prodrug of 6- pencylovir ann acylic guanosine analog. Similar m.o.A as acyclovir.The side chain of 6- pencyclovir differs from
that of acyclovir in that oxygen has been replaced by carbon and an additional hydroxymethyl grp.
M.o.A
Inhibit viral DNA sythesis. In HSV or VZV infected cells, penciclovir is initially phosphorylated by viral thymidine kinase.
Penciclovir triphosphate serves as a competitive inhibitor of DNA polymerase
Although penciclovir triphosphate is 100-fold less potent in ihibiting viral DNA polymerase than acyclovir triphosphate, its present in much higher
concentrations for more prolonged periods in infected cells than acyclovir triphosphate (7-20hrs) and this is associated with prolonged antiviral
effects.
Because it has 3’-hydrxyl grp penciclovir is not an obligate chain terminator but it does inhibit DNA elongation.
Clinical use
Treatment and suppression of recurrent genital herps and oral in immunocompetent individual
Treatment of oralabial and genital HSV infection in immunocopromised .
It s active invitro against HSV1, HSV, VZV, EBV and hep B
Adverse effects
Oral famiciclovir is well tolerated, though headache, diarrhoea, and nausea may occur.
Testicular toxicity has been demonstrated I animals receiving repeated doses, though men receiving daily famiciclovir (250mg every 12 hrs) had no
changes in spermatic morphology or motility.
Resistance
Currenly low
Can be due to thymidine kinase deficiency.
Acyclovir kinase – resistant herpes viruses are cross resistant to penciclovir.
Pharmacokinetics
Available as oral formulation
Bioavailability from oral admin is 70% of famciclovir.well and rapidly absorbed from intestine Food slows absortion but does not reduce ovaral
bioavailability.
Rapidly converted to pencyclovir.
Less than 20% is plasma bound.
A peak plasma conc 2µg/ml is achieved after following a 250mg oral dose.
Penciclovir triphosphate has a serum t ½ of 10 hrs in HSV-1 and 20 hrs in HSV-2.
Non renal clearance (mainly fecal) accounts for about 10%. Because penciclovir is excreted primarily in urine clearance if reduced in renal
insufficiency and dosage adjustment is required.
The plasma t ½ averages 2hrs and over 90% is excreted unchanged in urine.
The plasma t ½ averages 9.9 hrs in renal insufficiency. Haemodialysis efficiently removes penciclovir.
Penciclovir
Active metabolite of famciclovir.
Pharmacokinetics
Oral penciclovir has low(5%) bioavailability.
Used against HSV-1 and -2 and VZV, Penciclovir is similar in action to acyclovir, that is it is a chain terminator. It can only be used as a topical
cream because of insolubility
Topical application of 1% penciclovir cream has been shown to be effective for the treatment of recurrent herp labialis.
Ganciclovir
Acyc guanosine analog
This drug is very similar to Acyclovir, it just has an extra -OH. It is also available as a pro-drug called Valganciclovir which is an L-valine ester of Ganciclovir (Valcyte).
Oral Valganciclovir will probably to replace intravenous Ganciclovir for therapy and prevention of cytomegalovirus (CMV) infections. Ganciclovir is
active against CMV for which it is the drug of choice. Acyclovir has some activity against CMV in culture but has not found much use in therapy of these infections
because of the superiority of Ganciclovir.
M.o.A
As with Acyclovir, Ganciclovir targets the viral DNA polymerase and acts as a chain terminator. In herpes virus-infected cells, it is phosphorylated
first by the viral thymidine kinase and then by cell kinases to yield the triphospho form of the drug which is incorporated into and terminates the
DNA chain. However, CMV does not encode a thymidine kinase. Instead, Ganciclovir is phosphorylated by a CMV-encoded protein kinase (UL97) which accounts
for its specificity for infected cells. Selectivity is also achieved because the viral polymerase has 30 times greater affinity for Ganciclovir than the host enzyme.
Parmacokinetics
Ganciclovir is available in intravenous and oral formulations.The oral bioavailability of ganciclovir is poor (6-9%)cerebrospinal fluid conc. Valganciclovir, the oral
prodrug of ganciclovir, has been demonstrated equivalent to iv ganciclovir for CMV disease treatment in solid organ transplant recipients are approx
50%of those in serum. The t ½ is 2-4 hrs with normal renal function. Clearance of the drug is linearly related to creatinine clearance. Ganciclovir is readily cleared by
hemodialysis.
Clinical uses
Sight-threatening CMV retinitis in severely
immunocompromised people
CMV pneumonitis in bone marrow transplant recipients
Prevention of CMV disease in bone marrow and solid
organ transplant recipients
Confirmed CMV retinitis in people with AIDS (intravitreal
implant)
It is also used for acute CMV colitis in HIV/AIDS and CMV pneumonitis in immunosuppressed patients
Cytomegalovirus (CMV) used to rank first as a cause for morbidity and mortality among solid organ transplants (SOT) recipients Intravenous
administration of ganciclovir reduces the incidence of symptomatic CMV disease if admin before organ transplant
Normally, Ganciclovir is given intra-venously at a level of 10mg/kg per day or orally at 3000mg/day. It is often used for CMV retinitis in AIDS patients for
whom there is an intraocular (that is, intravitreal) implant known as Vitrasert. This contains 4.5 mg Ganciclovir for localized therapy.
Dual therapy with foscarnet and ganciclovir has been shown to be more effective in delaying progression rhinitis than either drug administered alone.
Intravenous ganciclovir is also used to treat CMV colitis and esopharigitis in AIDs PTs
It used to treat CMV pneumonitis particularly in combination with intravenous cytomegalovirus immunoglobulin.
Oral ganciclovir prevent end organ CMV disease in AIDs pts
Ganciclovir may also be administered by direct intravitreal injection.( an intraocular ganciclovir implant, which releases ganciclovir into vitreous cavity. This achieves
high prolonged intraocular levels of ganciclovir and has been shown to delay progression of retinitis).
A recent study showed that the combination of either IV or oral with intrvitreal in aids pts with CMV rhinitis resulted in a decreased incidence of Kaposi sarcoma over
6 months compared those receiving the inplant alone.
Resistance
Compartmentalized CMV disease refers to clinical syndromes wherein the virus is detected in the affected tissues but is minimally detectable or
undetectable in the blood. In the current era, gastrointestinal CMV disease (in the form of gastritis, esophagitis, enteritis, colitis) constitutes the vast
majority of tissue-invasive patients, and in a number of cases, this type of CMV disease is “compartmentalized.” Such a clinical presentation is
reminiscent of CMV retinitis, a very rare manifestation of tissue-invasive CMV disease after transplantation, that is often not accompanied by
viremia. This dilemma brings to the forefront the limitation of viral load monitoring in assessing duration of treatment.
Ganciclovir-resistant CMV is now emerging as an important complication of prolonged antiviral drug use after transplantation. Currently,
ganciclovir-resistant CMV is very rarely seen in liver transplant recipients (it is more common after kidney-pancreas and lung transplantation).
Unlike lung and kidney-pancreas transplant recipients who have rates as high as 9% and 13%, respectively, the estimated incidence of ganciclovir
resistant CMV after liver transplantation is < 0.5%. Several studies have identified risk factors for ganciclovir-resistant CMV, including CMV D+/Rstatus, high levels of viral replication, potent immunosuppressive therapy, and suboptimal ganciclovir levels.
Resistance to ganciclovir has been associated with mutations in the viral UL97 and UL54 genes
The former encodes a kinase that phosphorylates the drug to its monophosphate form, and the latter encodes the viral DNA polymerase that accepts
the triphosphate form of this compound.
Adverse effects
Low ganciclovir plasma concentrations have been associated with treatment failure and high concentrations with haematotoxicity and neurotoxicity,
but no formal therapeutic or toxic ranges have been validated.
Myelosuppression, particularly neutropenia
Myelosuppression may be addititive in pts receiving both ganciclovir and zidovudine.
Its also lower in oral than IV ganciclovir.
CNV toxicity headache, changes in mental status,
Ganciclovir is commonly associated with a range of serious haematological adverse effects. Common
adverse drug reactions (_1% of patients) include: granulocytopenia, neutropenia, anaemia,
thrombocytopenia, fever, nausea, vomiting, dyspepsia, diarrhoea, abdominal pain, flatulence, anorexia,
raised liver enzymes, headache, confusion, hallucination, seizures, pain and phlebitis at injection site
(due to high pH), sweating, rash, itch, increased serum creatinine and blood urea concentrations.
Ganciclovir is considered a potential human carcinogen, teratogen, and mutagen. It is also considered
likely to cause inhibition of spermatogenesis. Thus, it is used judiciously and handled as a cytotoxic drug
in the clinical setting.
Valganciclovir
Valganciclovir hydrochloride is a hydrochloride salt of the L-valyl ester of ganciclovir. It is a prodrug of ganciclovir.
Pharmacokinetics
Oral valganciclovir at the standard dose of 900 mg once a day produces a daily systemic exposure which is 1.7-fold greater than that attained with
oral ganciclovir at the standard dose of 1000 mg three times daily. In addition, the area under curve at 24 hours/drug exposure (AUC24) of
valganciclovir increased from 24% to 56% when compared from fed state to the fasting state.As a result, valganciclovir has greater CMVsuppressing ability than ganciclovir.
The absolute bioavailability of oral valganciclovir is approximately 60%, which is 10 times higher than ganciclovir bioavailability after oral
administration The peptide-mediated active transport of valganciclovir due to the presence of valine moiety explains its increased bioavailability. A
dose of 900 mg/day of valganciclovir produces a daily exposure equivalent to the 5 mg/day daily intravenous dose of ganciclovir. Oral ganciclovir
was developed as an initial attempt to provide a more convenient therapeutic regimen for ganciclovir (Cytovene ®; Roche Laboratories), but was
found to have low and variable bioavailability (6%–9%) compared to oral valganciclovir (60%). In clinical trials oral ganciclovir was found to be
inferior to intravenous ganciclovir for the induction phase, as it reached a subtherapeutic serum level. It was however approved by the FDA for
maintenance phase therapy of CMV retinitis.
Upon absorption, it is extensively and rapidly hydrolyzed into the active forms of ganciclovir, which is a mixture of two diastereomers, by hepatic
and gut esterases. Hence the pharmacokinetics of oral valganciclovir is essentially the same as that of intravenous ganciclovir.
Protein binding of valganciclovir has not been established due to rapid and extensive conversion of oral valganciclovir to ganciclovir. Kidneys
excrete valganciclovir as ganciclovir, both by glomerular filtration and active tubular secretion. Hence the plasma level of ganciclovir and thus
terminal half-life is increased in patients with renal impairment. Pharmacokinetic parameters are described in
Mechanism of valganciclovir
Ganciclovir is the only metabolite of valganciclovir and it undergoes no further metabolism. Intracellular phosphorylation of ganciclovir forms
ganciclovir triphosphate, which is the most active form. Ganciclovir triphosphate inhibits viral DNA synthesis by competing with deoxyguanosine.
This prevents the incorporation of deoxyguanosine into elongating viral DNA, thus terminating DNA synthesis, chiefly of the group of herpes viruses.
Ganciclovir monophosphate formed after the release of pyrophosphate is also incorporated into the growing viral DNA chain further slowing the
process of replication.
Resistannce to valgancliclovir
Viral resistance to ganciclovir has been noted with prolonged treatment in patients with CMV retinitis and was first reported in 1989. Ganciclovir
resistance has been attributed to the mutations in protein kinase gene UL97 and polymerase gene UL54. The incidence of ganciclovir resistance in
valganciclovir therapy is similar to that in intravenous ganciclovir therapy.
Mutation in viral protein kinase gene UL97
Deletion of four amino acids in a highly conserved region of viral protein kinase results in a mutation of UL97, viral protein kinase gene. These viral
mutants fail to phosphorylate ganciclovir efficiently and this is the main cause for CMV resistance to ganciclovir.
Mutation in viral polymerase gene UL54
Mutation in viral polymerase gene UL54 has resulted in cross-resistance with other antiviral drugs acting on viral DNA polymerase such as cidofovir
and foscarnet.This leads to reduced incorporation of the drug into the CMV DNA strand.
Adverse effects
General
The common adverse events reported during maintenance therapy are diarrhea, nausea, vomiting, abdominal pain, fever, headache, insomnia,
tremors, peripheral neuropathy, seizures, confusion, paresthesia, hallucinations, graft rejection, constipation, back pain, high blood pressure and
retinal detachment.
Myelosuppressant
Less than 5% patients had serious adverse reactions such as pancytopenia and aplastic anemia.
Teratogenic
Valganciclovir is a FDA pregnancy category-C drug with reproductive toxicity similar to ganciclovir. Common teratogenic abnormalities reported in
animal models include cleft palate, hydrocephaly, microphthalmos and brachygnathia. Adequate contraception is advised to women of reproductive
potential on valganciclovir.
Carcinogenic
Although there is no human study to prove the carcinogenesis of valganciclovir, patients are warned to consider it a potential carcinogenic.
Anti-fertility
It causes azoospermia in animals due to direct cessation of spermatogenesis, without affecting testicular endocrine functioning. Suppressed fertility is
reported in females in a different animal study.
Special precautions
Pediatric and geriatric population
The effects of valganciclovir on pediatric and geriatric population have not been studied. Caution is to be exercised while using valganciclovir in the
elderly keeping in mind the increased frequency of impaired renal, hepatic and cardiac function in them.
Nursing mothers
It is not known whether ganciclovir is secreted in human milk. Nursing mothers are instructed not to breast-feed if they are on valganciclovir. The
Centers for Disease Control also recommend HIV-infected mothers not to breast feed their infants to avoid postnatal transmission of HIV.
Contraindications
Valganciclovir is contraindicated in patients with ganciclovir hypersensitivity. It is contraindicated in patients with absolute neutrophil count less
than 500/mm3, platelet count less than 25,000/mm3 or the hemoglobin less than 8 g/dL.
Valganciclovir is not advised in patients on hemodialysis or in those with creatinine clearance below 10 ml/minute, as the optimal daily dosage is less
than 450 mg. There is no study regarding the effects of peritoneal dialysis on the excretion of valganciclovir.
Drug economics
Oral valganciclovir is an efficient and advantageous substitute for ganciclovir in many ways. It is an effective alternative to intravenous ganciclovir
in induction and maintenance therapy of CMV retinitis in HIV patients, and is found to achieve significantly higher systemic levels than oral
ganciclovir.
Valganciclovir is cost and time effective as it eliminates the expenses involved in hospitalization, intravenous, or indwelling catheterizations and
nursing care associated with the use of parenteral ganciclovir, foscarnet or cidofovir.
Long-term intravenous therapy predisposes the patient to the risk of catheter-related complications such as thrombophlebitis, intravenous line
infection and sepsis. Although valganciclovir is more expensive per pill than ganciclovir, the costs are comparable with intravenous ganciclovir
taking into account the cost of parenteral administration.Valganciclovir has overcome the large pill burden of oral ganciclovir and has improved
treatment adherence.
Cidoforvir
Chemical name: 1-[( S )-3-hydroxy-2-(phosphonomethoxy)propyl]cytosine dihydrate (HPMPC)
Other names: Vistide®
M.oA
Cidofovir is both a DNA chain terminator and DNA polymerase inhibitor. It is an acyclic nucleoside phosphonate (not a phosphate) in which the CO-P bond in a nucleoside monophosphate has been replaced by a phosphonate (C-P) bond that provides an enzymatically stable derivative with a
long half life.The drug is administered in the phosphonomethoxy-nucleoside form and is phosphorylated twice intracellularly to the active
diphosphate form using two cellular kinases (pyrimidine nucleoside monophosphate kinase and pyrimidine nucleoside diphosphate kinase. A viral
kinase is not involved, in contrast to acyclovir which is administered as the nucleoside form and the first phosphate is added by viral thymidine
kinase). Cidofovir inhibits the DNA polymerases of a number of viruses at concentrations that are substantially lower than those needed to inhibit
human DNA polymerases.
Pharmacokinetics
Although terminal t ½ of cidofovir is about 2.6 hrs, the active metabolite cidofovir diphosphate, has prolonged intracellular t ½ of 17-65 hrs.
Aseparte metabolite cidofovir phosphocholine has a halflife of atleast 87 hrs and may serve as an intracellular reservoir of active drug.
Elimination involves active renal tubular secretions.
Clinical uses
Treatment of CMV rhinitis
Treatment of polyoma virus associated progressive multifuctorial leukoencephalopathy
It may be useful for treatment of acyclovir-resistant herpes infections. It is also active against pox viruses, including the molluscum contagiosum
virus, BK virus, which is a polyoma virus, and adenoviruses. It is promising for the treatment of immunocompromised patients for gasteroenteritis
caused by adenovirus, although no control studies have been carried out, and has been used as an adjunctive treatment in addition to HAART in the
treatment of AIDS patients with progressive multifocal leukoencephalopathy (PML). The latter is caused by JC, another human polyoma virus.
Resistance
Due point mutation in DNA polymerase(K513N). Cross resistance the polymerase mutants associaterd with the longterm ganciclovir therapy may
occur.
No cross resistance to forscarnet has been reported.
Adverse effects
Dose dependent nephrotoxicity- serum creatnine and urine protein must be monitored within 48 hrs before each iffusion.
Concurrent admin of other agents with potentinal nephrotoxicity( amphotericin B, non-steroidal anti-inflammatory drugs ) should be avoided.
Other potential side effects include hypersensitivity reaction, ocular hypotony, uveitis.
Foscarnet
Foscanet( phosphomic acid)is an inorganic pyrophosphate compound that inhibit viral DNA polymerase, RNA
polymerase, and HIVreverse transcriptase directly, without requiring activation by phosphorylation. It has
invitro activity against HSV, VZZV, CMV, EBV, HHV-6, HHV-8 anddd HIV.
Pharmacokinetics
The drug is available in IV formulation.
Poor oral bioavailability and GI intolerencee preclude oral use. CSF concentrations are 43-67% of steady state
serum concentrations.
The mean plasma t ½ is 4.5-6.888 hrs. Up to 30% of the drug may be deposited in the bone, with t ½ of several
months. The clinical repercussions of this are unknown.
Clearance of foscarnet is primarily by the kidney and directly proportionate to creatine clearance. Serum drug
concentrations are reduced by approximately 50% by a 3-hour hemodialysis.
Clinical uses
Treatment of pts with CMV retinitis and acyclovir resistant HSV
Treatment of CMV colitis and esophagitis and acyclovir resistant VZV infection
Combination of forscarnet and ganciclovir has been shown tobe synergistic in the delay progression of
retinitis( the decision to administer combination therapy needs to take into account the severity of retinitis ie
whether potentially sight threatening or not as well as the potential negative impact on quality of life conferred
by the prolonged infusion times required to administer both drugs each day.
Forscarnet has been administered intravitreally for the treatment of CMV retinitis in Pts with AIDS.
Adecrease in the incidence of Kaposi sarcoma has been observed in Pts who have received forscarnet. However
treatment of Pts with Kaposi sarcoma using antiherpes agents has not been successful.
Resistance
Mutations that confer resistance to foscarnet map to different conserved regions in the UL54 gene and do not
appear to cluster around a specific site
Clinical data have shown that resistance to foscarnet is associated with mutations T700A, M715V, E756Q,
V781I, V787L, L802M, A809V, V812L, and T821I . Of note, it has been reported that prolonged treatment
with ganciclovir and forscarnet can lead to the emergence of a dually resistant phenotype. One particular isolate
harbored mutations T821I and K805Q among others
Adverse reactions
Renal insufficiency
Hypo- or hypercalcemia, hypo- or hyperphosphatemia
Penile alcerations associated with foscarnet therapy may be due to high levels of ionized drug in the urine.
CNS toxicities include: headache, hallucinations and seizures.
Use of an infusion pump to control the rate of infusion is important to avoid toxicity and relatively large
volumes of fluid are required because of the drug poor solubility. Saline preloading helps to prevent
nephrotoxicity, as does avoidance of conncomintant administration of drugs with nephrotoxic potential.
Concurrent adminstaion with pentamidine may exacerbate both nephrotoxicity and hypocalcemia.
Fomivirsen
Its aN oligonuCleotide that inhibits human CMV through an antisense mechanism. Binding of fomivirsen to
target mRNA muthoni
ANTI-INFLUENZA AGENTS
Zanamivir (Relenza) and Oseltamivir (Tamiflu)
Two neuraminidase inhibitors have been approved by the FDA (Zanamivir [Relenza] and Oseltamivir
[Tamiflu]). They are active against both influenza A and influenza B and can reduce the duration of
uncomplicated influenza (by approximately 1day in about 70-90% of adults) if taken within two days of the
onset of illness. However, oseltamivir resistance has been seen in some circulating strains recently (the 2009
H1N1 strain is sensitive).
To date there are only a few studies of how effective these drugs are in reducing serious complications in high
risk groups when used to treat influenza (as contrasted with when used prophylactically). Some limited data
suggest they may be beneficial. However, both are approved for prophylaxis as well as treatment.
M.O.A
Neuraminidase is an essential viral glycoprot for virus rep and release so the drugs inhibit neuraminidase.
Pharmacokinetics
Zanavir is admin intranasally. The cpn has poor oral bioavailability, limited plasm prot binding rapid renal
clearance and absence of significant metabolism
Oseltamivir is a prodrug that is given orallyand activated in the gut and liver. Its t ½ is 6-10hrs and excretion if
primarily in urine
Resistance
Due to mutation though the incidence of mutation are not known.
Rimantadine and amantadine
Rimantadine and amantadine block virus entry across the endosome and also interfere with virus release. They
may be given as protective agents during an outbreak, especially to those at severe risk and key personnel.
These drugs were widely used. However, in the 2005-2006 influenza season 92% of the H3N2 strains examined
had a mutation which would confer resistance to these drugs, as did 25% of the H1N1 strains tested - similar
problems have been seen in seasons since then so these drugs are not recommended until the level of resistance
in the major circulating strains drops. In the absence of the resistant mutations they were good prophylactic
agents for influenza A (but not for influenza B), although there are some problems in taking them on a long
term basis. They could be given to protect during an outbreak - especially those at severe risk and key
personnel. They could also be given at the time of vaccination for a few weeks - until the humoral response had
time to develop. There is some evidence that rimantidine and amantadine can reduce the duration of nonresistant influenza A if given early in infection.
M.O.A
The target is M2 protein within the membrane of Influenza A. Influenza B contains different protein.
Resistance
Rapid in treated individuals due to mutation. Transmission of resistant virus to household contact has been
documented.
Adverse effects
Gastrointestinal intolerance
CNS complaints (nervous, difficult in concentrating, lightheadedness)-less frequent with rimantadine than
amantadine.
Daily dossageshould be reduced in PTs with renal insufficiency and those over 65 yrs
OTHER ANTIVIRALS
Ribavirin
Interferons
gr
Palivizumab