1
Stable Isotope Geochemistry
Definitions:
Stable vs. radioactive or radiogenic isotopes
Stable isotopes that are used in geology: Isotopes that are used in geology all have large
relative mass differences (i.e. in relation to their common isotopes). This is the most
important criterion that makes them useful in geology. To explain this, the relative
difference in mass of the two O isotopes (18O and 16O) is 18/16 = 1.125 or 12.5%. The
relative mass difference for the isotopes of W (186W and 180W) is 186/180 = 1.03 or only
3%. Therefore, the oxygen isotopes are more useful than W ones. Isotopes of the light
elements H, C, N, O, Si, and S are therefore the ones used in geology. In addition to their
low atomic mass, and large relative mass difference, these isotopes have the following
characteristics:
1- They usually form bonds with a high covalent character (i.e., they are
electronegative elements).
2- Many exist in more than one oxidation state (e.g. C, N, and S)
3- The abundance of their rare isotope is still high enough to ensure accurate and
precise determination of the isotopic ratios.
4- They are important constituents of natural substances, forming a variety of
compounds.
Isotopic fractionation
Is the natural process by which the different stable isotopes of an element are separated
from one another.
Mechanisms of isotopic fractionation:
A- Physical non-equilibrium processes
1- Evaporation
2- Precipitation
3- Bacterial activity (often acting as catalysts for certain chemical reactions, increasing
the rate of chemical reaction. Unidirectional processes).
B- Equilibrium processes:
Isotopic exchange between coexisting phases to reach isotopic equilibrium. This
fractionation is influenced by many factors (see below), but in general, the heavier
isotope will be more fractionated in the compound or mineral with those bonds
having the stronger covalent character.
Example for an isotopic exchange reaction:
2 Si18O2 + Fe316O4 = 2 Si16O2 + Fe318O4
Gr for this reaction is very small (~ 1000 times smaller than bond energies or heats
of common chemical reactions), which makes isotopic equilibration a very slow
process. Therefore, if two minerals are in isotopic equilibrium, they must have
attained chemical equilibrium earlier! (Good test for chemical equilibrium). The
equilibrium constant for this reaction "K" is expressed as
K = [Si16O2 ]2 [Fe318O4]/[Si18O2 ]2 [Fe316O4]
2
Note that the activity coefficient values for these minerals () have been ignored,
which is common-practice in such calculations as they tend to cancel out.
The fractionation factor "":
Is the ratio of concentration of the heavy to light isotope in one phase divided by the same
ratio in the other phase of interest. Therefore, for any two phases "A" & "B", For any two
minerals A and B, and considering the fractionation of the isotopes of oxygen, is given
by the relationship
A-B= (18O/16O)A/(18O/16O)B
It can be seen that is directly proportional to the equilibrium constant "K".
Factors affecting "":
1- Temperature: As T increases, approaches unity. At very high T, stable isotopes are
not fractionated between coexisting phases.
2- Chemical bonding: The type of bond involving the element in question plays a major
role in controlling the value of . At equilibrium, the heavier isotope is more
concentrated in the phase with the stronger or more covalent bonds, and to the element
with the higher ionic potential. For example, 18O is more concentrated in quartz than in
coexisting magnetite, since the Fe2+ - O bond is much weaker (and more ionic) than the
Si - O bond. Similarly, 18O is more concentrated in calcite compared to witherite
(BaCO3) (cf. the electronegativity values of Ba and Ca), and 34S is more concentrated in
sphalerite (ZnS) than coexisting galena (PbS; cf. their ionic potentials and
electronegativity values).
3- Crystal structure: Has a secondary or minor effect on the values of . The heavy
isotopes concentrate in the more closely packed or ordered structure. Therefore, 18O is
more concentrated in ice compared to water, and in aragonite compared to calcite.
4- Pressure: Has a minor effect on isotopic fractionation. This is simply because the
change in reaction volume for an isotope exchange reaction is almost equal to 0 (Vr =
0). However, in the case of exchange between a fluid and a solid, P may play a more
significant role in affecting .
Analysis for stable isotopes:
Extraction
Conversion to a gas (CO2, H2O, SO2 or SF6, ... etc.)
Mass spectrometry and isotopic ratios
The stable isotopes of Oxygen and Hydrogen:
Expression of Isotopic ratios:
Isotopic ratios of a mineral are compared to those of a standard, and expressed in the
"delta" notation. For example, in the case of oxygen isotopes 18O is given by the
relation:
18O = 1000.[(18O/16O)A - (18O/16O)SMOW]/(18O/16O)SMOW %
°
where SMOW stands for standard mean oceanic water, the standard to which isotope
ratios are compared. Note that the value of 18O is expressed in "per mil" rather than %,
3
since it is too small. Obviously, 18O of the standard SMOW will be zero. It is also clear
that
A-B = (1000 + 18OA)/(1000 + 18OB)
or
1000 ln A-B A - B
The difference between the values of two minerals is denoted by "A-B".
Applications:
1- Geothermometry:
Consider the reaction:
2 Si18O2 + Fe316O4 = 2 Si16O2 + Fe318O4
in which the stable isotopes of oxygen (18O and 16O) are being exchanged between the
phases quartz and magnetite. The equilibrium constant "K" for this reaction is therefore
directly related to the ratios of both isotopes in magnetite and quartz , and is a function of
T. Although Gr is very small, the fact that K (or !) are insensitive to P (since Vr of
stable isotope exchange reactions is virtually zero), makes this exchange a useful
geothermometer, especially if the fractionation of the oxygen isotopes between these two
minerals is fairly large (due to differences in bond strengths or large structural
differences) and if they equilibrated at low T. Based on chemical bond strength, common
minerals can be arranged according to their tendency to concentrate 18O as follows:
Qz > K-spar ~ Arag ~ Cc > Musc > An ~ Gln > Lw > Cpx > Gt > Bt > Ol > Chl > Rt >
Ilm > Mgt.
According to this list, mineral pairs that are most suitable for isotope thermometry (ones
characterized by the largest differences in 18O values) are: quartz - rutile, quartzmagnetite, and feldspar - magnetite. On the other hand, although it may be tempting to
use the quartz - calcite pair for the determination of the temperatures of metamorphism of
siliceous carbonates, fractionation between these two minerals is not very sensitive to T
changes. Moreover, the isotopic composition of this pair is easily reset during
retrogression, or access to fluids.
Since "" is independent of bulk rock compositional variations, and is also not strongly
affected by pressure or solid solution in the minerals used (except where such solid
solution causes a change in the bond strength), O-isotope geothermometry should
therefore be an ideal method for temperature determination. Theoretically, if a rock
consists of n phases in equilibrium, it would have n-1 isotopic geothermometers that
should all give the same T of equilibration. The T dependence of is expressed as:
103ln = (106. A/T2 )+ B
where A & B are two constants that depend on the types of minerals used, and their
structures. This relationship is shown in Fig. 1. Values for A and B are given by Bottinga
and Javoy (1975), and several papers by Matthews (e.g. Matthews, 1994).
However, O-isotope geothermometry suffers from the ease with which some systems are
reset by retrogressive processes, with the 18O values of minerals strongly influenced by
the last fluid with which they have equilibrated. Moreover, exchange between many pairs
4
has not been calibrated in the laboratory, and changes in the strength of some bonds as a
result of some types of solid solution add a further complication, as more experiments are
needed to cover larger compositional ranges. The tedious process of mineral separation
has also limited the use of this method, although this is now changing with the advent of
laser techniques.
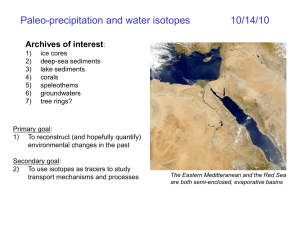
2- Tracing the origins of the different types of “waters”:
D - 18O plots (Figs. 2 - 5),
Meteoric water line and the origin of hydrothermal mineral deposits (Fig. 5).
3- Identifying the types of fluids with which the rocks may have interacted
This is a simple outgrowth of applications 1 & 2. If has been determined that a rock has
interacted with a fluid in some way, then the isotopic signature of that rock should reflect
such interaction. For example, rocks that have been affected by hydrothermal alteration
with meteoric waters will have lower 18O values compared to those affected by
metamorphic or magmatic fluids (Figs. 3 & 4).
4- Testing for chemical equilibrium (Fig. 6)
If minerals separated from a rock have 18O values consistent with those predicted at a
given T on the basis of theoretical considerations (see the sequence of 18O values listed
above under geothermometry), then the likelihood that these minerals are in equilibrium
is very high. On the other hand, irregularities in this sequence (known as isotopic
reversals) are clear evidence for lack of equilibration. Note that if a system is in isotopic
equilibrium, it will definitely be in chemical equilibrium!
5- Calculation of fluid : rock ratios:
For closed system infiltration of rocks by a fluid (i.e. the situation where all fluid
simultaneously equilibrates with the rock), the fluid : rock ratios are calculated using the
following formulation:
F/R = f(rock) - i(rock)/i(fluid) - f(fluid)
where F and R are the atom percentages of the element of interest in the fluid and rock (in
this case O), i and f are the initial and final values of rock or fluid, respectively. In
practice, f(rock) is measured, i(rock) can be measured if we can find rocks that have
not been infiltrated by this fluid, i(fluid) is assumed, whereas f(fluid) can be calculated
if the T of infiltration is known (from experimentally determined fractionation factors).
6- Climatological and paleoclimatological studies:
Fractionation of the isotopes of O and H between ice, water and vapor: 18O and
D increase from vapor to ice (Fig. 7a & b).
Although evaporation is not an equilibrium process, condensation is.
As the T decreases, the fractionation of the isotopes also increases
Lowest (most negative) values of 18O and D are recorded for rainwater and
snow in polar areas (Figs. 8 & 9).
Rationale could be used to investigate the origins and sources of groundwaters in
various aquifers (Fig. 10), and for paleothermometry.
5
Sulfur isotopes
Ratio is that of 34S/32S
Standard is troilite (FeS) in a meteorite from Arizona
34S ranges from -30 to +60 %° (Fig. 11)
34S controlled by oxidation reduction reactions and the exchange of S between
sulfides and sulfates.
Other factors affecting 34S :
1- pH
2- fO2
3- T
4- Reaction rates & bacterial activity
Carbon isotopes:
Ratio is that of 13C/12C
Standard is that of calcite from a belemnite from Peedee Formation in South
Carolina (PDB)
Oxidation and reduction are effective means for fractionation
13C is preferentially incorporated in CO relative to any organic compound (C-H
2
bonds). Photosynthesis therefore results in the concentration of the light isotope in
organic compounds. Low values of 13C reflect some organic origin.
 0
0