vxcbcvbcvb
advertisement

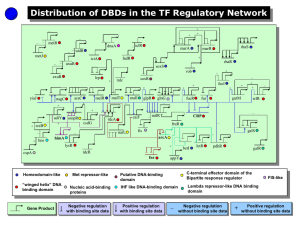
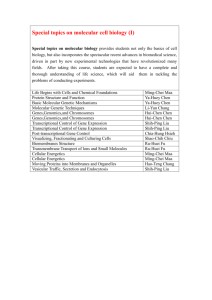
Summary The nature of an organism is determined largely by the dynamic interplay between its gene repertoire and the regulatory apparatus, which consists of transcription factors. The assembly of regulatory interactions linking transcription factors (TF) to their target genes (TG) forms a transcriptional regulatory network. This thesis focuses on the following fundamental aspects of evolution of transcriptional networks: (i) evolution of TFs, (ii) evolutionary mechanisms of network growth, (iii) types of changes during evolution, and (iv) dynamic response of network structure to different growth conditions within an organism. (i) A computational procedure that identified 271 putative TFs in the E. coli genome was developed. Evolutionary analysis of domain organisation of TFs revealed that the DNA binding domains (DBD) belong to one of the 11 SCOP superfamilies. We showed that ~75% of TFs evolved by gene duplication. It was also shown that proteins that contain DBDs from the same superfamily could have different regulatory functions. It is rather the position of TF binding site within DNA that determines its function as an activator or a repressor. (ii) Mechanisms for the evolution of regulatory networks were developed. Analysis of domain organisation of TFs and TGs in the known E. coli and yeast regulatory networks revealed that close to 90% of all regulatory interactions have evolved through duplication of the pre-existing genes. Regulatory interactions were either inherited or modified subsequent to gene duplication. We also showed that global and local structure of regulatory networks cannot be attributed to duplication alone, indicating the action of other evolutionary forces. (iii) A computational procedure that allowed reconstruction of transcriptional regulatory networks for 175 prokaryotic genomes was developed. Analysis of the reconstructed networks led to characterization of patterns of evolutionary change in both global and local network structure. We showed that regulatory hubs have been lost or replaced in the absence of apparent selective pressure to maintain them. Levels of conservation of regulatory motifs found within transcriptional networks were also assessed. An intriguing finding was that the same gene could be a part of different motifs in different genomes. This study provides the first overview of transcriptional control in poorly characterised genomes. (iv) For the yeast genome, gene expression data across different growth conditions was integrated with the known transcriptional network in order to identify condition specific sub-networks. We found that the structure of these sub-networks differed significantly. The set of regulatory hubs was different for various conditions, suggesting that differential expression of key hubs could induce particular cellular conditions. We discovered that different types of network motifs are being preferentially used in different conditions and provide insight into the dynamic usage of regulatory interactions within an organism. This thesis illuminates principles in the evolution of transcription factors, networks within & across organisms and the dynamic usage within an organism. It also provides predictions that could be useful in engineering regulatory networks in different organisms.