doc
advertisement

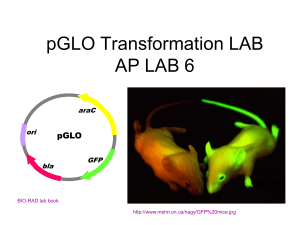
Notes for Preparation/Demonstration of Practical Session: Preparation and Transformation of Competent E. coli and Selection of Transformants. Aims and Objective of Practical: Preparation and transformation of competent cells is one of the fundamental techniques used in cloning regimes and this practical session trains students in fundamental methodology (preparation and transformation of competent cells; sterile technique; safe handling of micro organisms; serial dilution), use of basic equipment (centrifuges, pipetting) underlying principles (demonstrates principle of selection of transformants based on visible evidence of gene expression). Additional documents provided for use: Technical support and preparation notes Practical demonstrators introductory notes. Suggested organisation of students: Ideal for pairs or for individual work. Risk Assessment and Safety Considerations: UK version is given on front page of sample schedule. Requirements and regulations might vary from country to country and should be checked and modified accordingly before the practical is undertaken. Learning Outcomes for Students: Suggested at start of schedule. Assessment: The practical schedule can be assessed in several ways, and should be adapted depending on level of student, prior experience and learning outcomes of the programme of study in which the practical will be delivered e.g. either via a traditional ‘write up/report’, with data being collected during the practical session and written up later, or by a mixture of multiple choice questions and guided calculations etc. Assessment based on answers to multiple choice questions are given [ideal for large student cohorts]. A sample student practical schedule is attached. This should be adapted depending on level of student/ prior experience/ facilities available in lab etc. Time Required: Session fits in 3 hour slot. Technical support and preparation notes All plastic/ glassware should be autoclaved and dried before use, or purchased pre-sterilised. Water used should be double distilled and deionised. Solutions should be aliquoted and autoclaved or filter sterilised before use. Equipment: Amount / group Microcentrifuge -1 (per 2-4 groups, ideally) Whirlymixer -1 (per 2-4 groups, ideally) Waterbath (or heatblock/oven) -1 (per session) 37oC and 42oC Microcentrifuge tubes . ~5 Microcentrifuge rack 1 P1000 Gilson micropipettes (or equivalent) 1 (ideally) P200 “ “ “ 1 P20 “ “ “ 1 gilson micropipette tips box p1000&p200/20 / group Solid (tip) waste container -1 (can share) Liquid waste container -1 (can share) Stop clock 1 Indelible marker pen 1 Sterile serial dilution tray, or extra microcentrifuge tubes 1 (can share) Sterile plate spreaders 4 per group (if disposable) or 1 & flaming ethanol and a bunsen if non-disposable UV lamp 1 (per session) Temperature controlled growth cabinet 1 (per session) Autoclave or pressure cooker; or sterile filters [0.44/ 0.22 m for sterilising solutions] Ice bucket and crushed ice 1 (can share) 10% bleach v/v bleach solution or disinfectant (e.g. virkon) bottle (to share) Materials: o Pellet of ~10ml overnight culture [mid log phase] of DH5 grown in LB + ampicillin (or any equivalent ACPD category 1 bacteria- note that antibiotic required might change depending on baterial strain used)- 1 per group o TFB (transformation buffer); 50 mmol/l KCl, 45 mmol/l MnCl, 50 mmol/l CaCl2, pH 6.2 o DMSO: (dimethyl sulphoxide) o DTT (dithiothreitol); 2.25 M o 5 ml sterile double distilled and deionised water [autoclave or filter sterilise]. o TE buffer; 10mM Tris.Cl pH 7.5, 1mMEDTA pH8 o 4 LB agar & ampicillin plates and 4 LB agar & ampicillin and arabinose plates per student group (see below) o Luria broth (LB). For 1 l: 10 g tryptone, 5 g yeast extract, 10 g NaCl (RMM 58.44), make up in double distilled and deionised water and pH to 7.0. Autoclave for use. Small volumes may be filter sterilised. o LB agar plates: add 15 g agar per l of LB. Makes 30-40 plates. o Ampicillin, 10mg/ml. Make in double distilled and deionised water and filter sterilise. Do not autoclave. Store at –20 °C for up to 1 year. Use at a final concentration of 0.1 mg/ ml (i.e. 1 ml of 10 mg/ ml stock solution per 100 ml of media). Add once LB agar has been melted and cooled to <50 oC. Dry, inverted, at 37 oC. Can be stored at 4 oC for 1 month. o 10 l (containing at least10 ng) pGLO per group [or equivalent appropriate expression vector with cloned GFP [note that antibiotic/ inducer requirements might differ with different vectors used] Predicted Results: As given in the introduction to the practical o Growth of both P and C cells on the LB amp plates o No growth of C cells on lb AMP or LB AMP& ARA o Growth of P cells on lb AMP- but no colour under UV illumination o Growth of P cells on lb AMP&ARA- and fluoresce green under UV illumination Practical demonstrators introductory notes. Draw student attention to the hazards and safety considerations that they have to be aware of (detailed in the risk assessment box) before they start work Lab coats and glasses on at all times; Live cells being used- so nothing down sink; liquid and solid waste to be segregated and any spills disinfected. Note handwashing - at end of practical, and if contaminated with cells. Students must read and prepare ahead, note solutions need to be pre-chilled and warmed. Remind importance of sterile technique - sources of contamination, keep lids on tips etc. Use correct pipettes [p1000 for 200-1000l; p200 for 20-200l; p20 for <20l]. Remind how to set pipette correctly Draw attention to importance of fully resuspending the cell pellets Timing of steps important, especially heat shock step - too short and cells will not get transformed, too long and cells will die - do not guess, use stop clock. Outline organisation of session (numbers per group/ number of groups/ location of equipment etc.) Explain arrangements for collecting results at a later date once cells have had sufficient incubation time Explain assessment. [Suggested] assessment is a series of calculations and questions at the end of the schedule that should be completed (so, be aware of schedule when organising benchwork!) Note on copying and plagiarism: everyone works in groups and are welcome to discuss results, but answers but be an individuals own work! Run through essence of practical (detailed in schedule, so no need to go into fine points) Step 1: making competent cells Step 2: transformation of cells with pGlo plasmid Step 3: selection of transformants with antibiotic and regulation of expression of GFP protein with arabinose PREPARATION AND TRANSFORMATION OF COMPETENT E. coli AND SELECTION OF TRANSFORMANTS COSHH Risk Assessment and Safety Considerations Read the following before you begin your practical work: Escherichia coli: ACDP category 1 (minimum risk). Dispose of live cells and anything that has been in contact with them in the designated waste containers for autoclaving. Wash hands thoroughly after handling, or if contaminated, with anti- microbial detergent. Wipe up any spills with disinfectant. Dithiothreitol: Toxic/corrosive. Wear gloves and safety glasses. If exposed, flush affected area with water for at least 10 min and report to the demonstrator for medical attention. Dimethyl sulphoxide: Toxic/corrosive/flammable/carcinogenic. Wear gloves and safety glasses. If exposed, flush affected area with water for at least 10 min and report to the demonstrator for medical attention. Centrifugation: tubes must be balanced to within 0.1 g of each other and arranged in a balanced pattern within the centrifuge rotor before starting the centrifuge. UV light: high-energy radiation. Do not look directly at the UV light source or expose naked skin. Doing so may result in damage to your eyes and skin. Wear lab coats and safety glasses. Learning Outcomes: At the end of the practical session and directed learning you should be able to: Handle microorganisms using sterile technique Prepare competent E. coli Transform competent E. coli Calculate the number of transformants Discuss gene expression in microorganisms INTRODUCTION: In this practical session you will transform a strain of E. coli (called DH5) with a recombinant plasmid (called pGLO - see later). However, many strains of bacteria, including the one you will be using, are not naturally competent (i.e. naturally able to take up DNA), so you will first have to make competent cells and then transform them with the plasmid. Transformation is very inefficient, so there must also be a simple way of selecting transformants (cells which have taken up the plasmid). A common method is to use antibiotic resistance screening. DH5 cells are sensitive to the antibiotic ampicillin. Therefore, if you plate them onto agar medium containing ampicillin they cannot grow. Bacterial chromosom e Plate onto nutrient agar containing ampicillin DH5 NO GROWTH: untransformed cells are ampS Ampicillin has its mode of action through inhibiting cell wall biosynthesis; A vital component of bacterial cell walls is peptidoglycan (also called -lactam) and ampicillin possesses a lactam ring, which interferes with cell wall synthesis. pGLO has been engineered to include the antibiotic resistance gene, bla, which encodes the enzyme -lactamase and confers resistance to the antibiotic ampicillin. Thus, when DH5 cells are transformed with the plasmid pGLO they can grow on agar medium containing the antibiotic ampicillin. pGLO PLASMID which carries the bla gene Bacterial chromosome Plate onto nutrient agar containing ampicillin DH5transformed with pGLO GROWTH of transformed colonies: AmpR In addition, the Green Fluorescent Protein (GFP) cDNA from the bioluminescent jellyfish Aequorea victoria has been cloned into pGLO. The GFP cDNA in pGLO is under the control of the arabinose operon promoter sequence (pBAD) - a DNA “switch” that allows its expression to be regulated. The arabinose operon is a cluster of 3 genes (araB, araA and araD) that encode enzymes that metabolise arabinose and their expression is dependent on initiation of transcription from the pBAD promoter sequence. When arabinose is not present in the growth media, an inhibitory protein (araC) binds to pBAD and prevents transcription of the operon. When arabinose is present, it binds to the araC protein causing a conformational change and initiation of transcription. When all the arabinose has been metabolised araC returns to its original shape and transcription is shut off. The DNA code of pGLO has been engineered to include the araC gene and the pBAD promoter; However, the arabinose operon has been replaced by the GFP gene, therefore GFP can be produced, but only in the presence of arabinose. A diagram of the pGLO showing the 3 genes used for selection of transformants and regulation of GFP expression is shown below: (1) the bla gene; (2) the araC gene (3) the GFP gene ara C Regulates expression of GFP gene. + arabinose: allows GFP expression - arabinose-: binds pBAD and blocks expression pGLO lactamase Confers resistance to ampicilin. pBAD promoter Ampicillin: lactam ring inhibits cell wall synthesis GFP Inactive ampicillin: GROWTH Fluoresces bright green under UV light PREPARATION FOR PRACTICAL: please note 1. Success depends upon pre-chilling/pre-warming solutions. Read ahead and prepare in advance. 2. Timing of steps is important- use an accurate watch or stop clock 3. You are using live cells - nothing that has come into contact with the cells should go down the sink. [if you do forget, report it to a demonstrator so they can disinfect the sink] 4. Sterile technique is important- only use sterile tubes and tips (and keep the lids on!); be careful not to contaminate anything e.g. your culture plates, tips etc. with dust, fingers, hair etc. 5. You will need to assess your results after the practical session - the demonstrator will explain the arrangements. MATERIALS: The following procedure should enable you to transform E. coli using a plasmid extract such as you have/ will prepare(d) in a linked practical [see associated practical: Isolation of plasmid DNA and quantification using UV spectrophotometry]. However, an aliquot of fresh plasmid is provided for convenience. Due to the time constraints of the practical you are also provided with a centrifuged pellet of an overnight culture of E. coli, strain DH5. METHODS: 1. Place an empty labelled microcentrifuge tube, the sterile transformation buffer (TFB) and the microcentrifuge tubes containing 10 l plasmid solution and 10 l sterile TE buffer on ice while you read through the rest of the schedule and organise yourselves. 2. Fully resuspend the pellet of E. coli in 1 ml of pre-chilled TFB using the whirlymixer and/or by gentle pipetting (you should have a smooth creamy suspension, with no lumps - not even small ones!). Transfer the suspension to the pre-chilled microcentrifuge tube and incubate on ice for 3 min. 3. Making sure your tubes are balanced, centrifuge your tubes for 30 seconds in the microcentrifuge, in order to pellet the cells. 4. Remove the supernatant and fully resuspend the pellet in 880l of pre-chilled TFB. Microcentrifuge tube supernatant Cell pellet- FULLY RESUSPEND by vortexing and/or pipetting 5. Add 40 l of DMSO and gently pipette several times to mix. Incubate for 2 min on ice. 6. Add 40 l of DTT and gently pipette several times to mix; Incubate for 2 min on ice. 7. Add 40 l of DMSO and gently pipette several times to mix; Incubate for 5 min on ice. The cells should now be COMPETENT (i.e. able to take up DNA). 8. Add 500 l (i.e. half) of your competent cells to the pre-chilled pGLO plasmid extract. Label this sample e.g. as “P” 9. Add 500l (i.e. the other half) of your competent cells to the pre-chilled sterile TE buffer (which will act as a control). Label this sample e.g. as “C”. 10. Incubate both samples on ice for 10 min. 11. You are now going to HEAT-SHOCK BOTH samples at 42oC. [The timing of this step is critical - heat shock for too long and the cells will die, too short and the cells will not take up the DNA]. Incubate both of your cell suspensions at 42 ºC, with thorough, occasional mixing, for precisely 90 SECONDS. 12. Incubate on ice for 2 minutes. 13. Add 1 ml of sterile nutrient broth (NB) to BOTH P and C and incubate for at least 15 minutes at 37ºC [to allow the cells to recover from their heat shock treatment]. While your cells are incubating, organise and label the nutrient agar plates you will require: 14. In total - you will require2 LB agar plates, 4 LB plates containing ampicillin (LB-AMP) and 4 LB agar plates containing ampicillin and arabinose (LB- AMP & ARA) and they should be labeled on the base of each plate - [not the lid, which can get accidentally swapped!] according to the following table P cells experimental- transformed with pGLO plasmid LB Plates required LB- AMP LB-AMP&ARA 100 (undiluted) 100 (undiluted) 10-1 (1 in 10 dilution) 10-2 (1 in 100 dilution) 100 (undiluted) 10-1 (1 in 10 dilution) 10-2 (1 in 100 dilution) C cells control- ‘transformed’ with TE 100 (undiluted) 100 (undiluted) 100 (undiluted) 15. Prepare the serial decimal dilutions for your P cells as follows: i) Pipette 900 l sterile water into two separate wells of the sterile dilution tray ii) Mix your P cells using a whirlymixer iii) Add 100 l of your resuspended cells to 900 l of water in the first well. Mix by gentle pipetting - this is a 10-1 (1 in 10) dilution. iv) Change pipette tips and add 100 l of the 10-1 dilution into 900 l of water in the second well. Mix by pipetting- this is your 10-2 (1 in 100) dilution. 100l 100l Undiluted cells (100) 10-1 10-2 900l H20 16. Using a fresh sterile spreader for each plate (if you are unsure of the technique ask for a demonstration), spread out 100 l of each of your dilutions onto the labelled plates. 17. Incubate the plates inverted. CHECK YOUR PLATES FOR COLONY GROWTH/COUNTING AT A TIME SPECIFIED BY THE DEMONSTRATOR- you will need these results to answer the assessed questions. Results: Colony counts: NOTE: if your group did not get any results remember to ask the demonstrator for sample results to use for the assessment - Observe your C plates. Number of colonies:………………………………………………………………………….. Colony Morphology (e.g. colour, size etc.):………………………………………………… Appearance under UV light:…………………………………………………………….…… Other notes:…………………………………………………………………………………… ……………………………………………………………………………………….….……… - For P select the spread plate which contains 10-100 colonies for observation [usually the 10-1 dilution] Number of colonies:…………………………………………………………………………….. Colony Morphology (e.g. colour, size etc.):………………………………………………….. Appearance under UV light:…………………………………………………………………… Other notes:…………………………………………………………………………………….. …………………………………………………………………………………………………… - Check that the dilution series for P shows a stepwise decimal reduction in cell number (i.e. there should be 10x less colonies on each plate as you move from 10 0 to 10-1 to 10-2) Notes:…………………………………………………………………………………………… …………………………………………………………………………………………………… …………………………………………………………………………………………………… Assessment: 1.Who first demonstrated transformation? A. Mary Lyons B. F. griffith C. L. J. Alloway D. E. Tatum E. J. Watson 2. Why must E. coli be made competent in this experiment? A. To help them grow faster B. To make the serial dilutions of bacteria suspension more accurate C. Because S. pneumoniae cannot undergo transformation D. Because some species of bacteria (including E. coli) are not naturally competent to undergo transformation E. To make the plasmid integrate into the bacterial chromosome 3. During this experiment, some bacteria in your ‘P’ sample will not take up the plasmid. What will happen to the non- transformed bacteria when plated onto the LB-AMP agar? A. They will grow slowly and form a bacterial lawn during an overnight incubation B. They will grow more slowly than the control cells C. They will grow slowly, but colonies will be detected after an overnight incubation D. They will conjugate E. They will not grow 4. What is the mode of action of -lactamase, the bla gene product? A: It removes the -lactam ring from the antibiotic ampicillin. B: It interferes with bacterial cell wall biosynthesis. C: It interferes with bacterial cell division. D: It degrades antibiotics E: It removes the -lactam ring from the antibiotic tetracyclin. 5. Calcium chloride (CaCl2.2H20) has the formula weight (molecular weight) of 147.0. It is used in the transformation buffer at 50mM (50 millimoles per litre). How much CaCl 2 would you weigh out to prepare 1litre of transformation buffer? A: 7.35 g B: 73.5 g C: 735 g D: 7.35 mg E: 73.5 mg Use your own results (or if your experiment did not work use the sample results provided by the demonstrator) to answer questions 6, 7 and 8. Dilution of spread plate counted:______________ Number of colonies counted:__________________ 6: When calculating the number of transformed (blue) cells, what is the factor you should multiply the number of colonies counted by, in order to take into account the dilution of the spread plate you counted? A: 0 B: 1 C: 10 D: 100 E: 1000 7. When calculating the number of transformed cells per ml, what is the factor you should multiply the number of transformed colonies by, in order to take into account the volume of cells spread onto the plate you counted? A: 0 B: 1 C: 10 D: 100 E: 1000 8: What is the number of transformed cells per ml which you produced? --------------9. Explain your results in terms of the regulated expression of GFP in pGLO. Your answer should be concise and clear. ANSWERS. 1. A 2. D. 3. E 4. E 5. A 6. If 100: ANS:B; if 10-1: ANS: C; if 10-2: Ans: D 7. C 8: no of cells x 10 x ans from 6 – must be sensibly and well expressed in cells/ ml [cells ml-1] 9. Concise and clear explanation of bla gene product and interaction between araC gene product and pBAD promoter in presence/ absence of arabinose.