Abstract - Open Access Repository of Indian Theses
advertisement

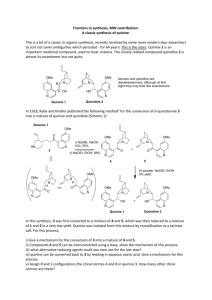
ABSTRACT The thesis entitled “Studies towards the synthesis of fumagillin & ovalicin and development of new synthetic methodologies” is divided into three chapters. CHAPTER I: This chapter is further divided into two sections. Section A : This section describes introduction and previous synthetic approaches for fumagillin. Section B : This section describes introduction and previous synthetic approaches for ovalicin. CHAPTER II: This chapter includes our contribution for the synthesis of fumagillin and ovalicin and is further divided into two sections. Section A : This section describes the synthesis of a key intermediate for the total synthesis of fumagillin . Section B : This section describes the formal total synthesis of ovalicin. CHAPTER III: This chapter deals with the development of new methodologies and is further divided into two sections Section A : This section describes introduction of α- amino phosphonates, α- hydroxyl amino phosphonates and previous approaches for the synthesis α- amino phosphonates Section B : This section describes the present work for the synthesis αamino phosphonates and α- hydroxyl amino phosphonates. 1 Abstract Chapter II. Scection–A: This section describes the stereo selective sythesis of a key intermediate for the total synthesis of fumagillin . In 1990 Judah Folkman and co-workers discovered that fumagillin 1 isolated from aspergilles fumigatus and its semi-synthetic drug TNP-470, 2 (also known as AGM 1470) have the remarkable capacity to inhibit the proliferation of endothelial cell in vitro and tumor induced angiogensis in vivo (Fig 1). Fumagillin and its less toxic semisynthetic derivative TNP-470 are potential antimalarial and anti-leishmaniasis drugs and are the only treatments for intestinal microsporidiosis in HIV-infected patients O O HO O O O OMe O N H OMe Cl O O O O Fumagillin-1 Me AGM 1470. 2 Fig 1 Retro synthetic strategy : The retro synthetic strategy was devised and outlined in Scheme 1. In this the main core cylohexane moiety 3 could be obtained from 4 by extending the aldehyde group. The compound 4 could be obtained from olefinic alcohol 5 which in turn could be synthesized from our well known ring opening reaction of alcohol 6. It was also envisioned that the alcohol compound 6 could be easily obtained from 7, which was accessed from the Diels-Alder reaction of protected furfuryl alcohol 8 and bromo methyl propiolate 9. 2 Abstract OT BS OH OMe OH OMe OMe CHO COOMe Fumagillin-1 O O O 3 OMe OMe O 5 4 Br O COOMe COOMe OH Br R=TBDMS R=PMB 8a-R=TBS 8b-R=PMB OR 7a-R=TBS 7b-R=PMB 6 + OR O COOM e 9 Scheme 1 Our first approach is outlined in Scheme 2. The Diels-Alder reaction of TBS protected furfuryl alcohol 8a and bromo meyhyl propiolate 9 in refluxing benzene gave the adducts 10 and 7a in 3:7 ratio. The regiochemistry of the two adducts was assigned from their 1H -NMR spectroscopy. The adduct 7a when treated with NaOMe in MeOH gave the dimethoxy compound 11 in good yields. Compound 11 on hydrogenation followed by TBS deprotection with TBAF in THF gave the alcohol 6. Where as the Diels-Alder reaction of PMB protected furfuryl alcohol 8b and bromo meyhyl propiolate 9 under similar conditions gave the adduct 7b as a single regioisomer. To check the regio chemistry of the adduct, we applied the same set of reactions as above and obtained the same alcohol 6. Scheme 2 COOMe Br O OTBS COOMe O Br COOMe OMe COOMe OTBS 11 OMe O COOMe THF,95% OH OTBS 6 OMe OMe Br NaOMe, MeOH O Benzene, reflux, OPMB 24 h, (58%). 1 h, (78%). 7a TBAF OMe COOMe 12 O OMe OMe 12 h, (93%) 8b COOMe OTBS 10 H2, Pd/C, MeOH NaOMe, MeOH O OTBS 8a O + Br Benzene, reflux, 24 h, (58%). OMe Br O COOMe O 1 h, (78%). COOMe OPMB OPMB 7b 13 3 OMe H2, Pd/C, MeOH OMe O 12 h, (93%) COOMe OH 6 Abstract The free 1o-alcohol was transformed to iodide using iodine, TPP and imidazole in refluxing toluene. This iodo compound 14 when treated with TMSOTf in DCM gave the keto compound 15. NaBH4 reduction followed by methylation resulted the iodo methoxy compound 17. Zn mediated ring opening reaction of iodo methoxy compound 17 in refluxing ethanol gave the olefinic alcohol 5. Protection of the sec-alcohol as TBS ether by using TBS-Cl followed by dihydroxylation of the double bond with OsO4-NMO resulted in diol 19. Acetonide protection of the diol followed by DIBAL-H reduction gave the aldehyde compound 21 which is in contrast to desired aldehyde 4 from the noe studies, which revealed that the two sec-hydroxyl groups were not in the same plane which is mandatory for the target molecule, instead the methoxy group was in the same plane with aldehyde (Scheme 3). OMe OMe I2 , TPP,IMD. OMe O toulene, ref, 2 h, (93%). COOMe O I 15 I 6 14 OH O MeI, Ag2 O. OMe O COOMe THF, 24 h, (87%). OTBS OH OMe Zn, Ethanol. COOMe ref, 2 h, (88%) OMe TBS-Cl, IMD. COOMe DCM, 1 h, (73%). COOMe I I 16 OsO4 ,NMO, Acetone:H2 O NaBH4 ,MeOH. COOMe r.t, 1 h, (80%). 0 0 C, 10 min, (60%) COOMe OH O TMSOTf, DCM OMe O 5 17 OMe OMe CSA, Acetone. O O 19 20 DIBAL-H, DCM 4 4 CHO CHO O O Scheme 3 OMe OMe COOMe -78 0 C, 1 h, (66%) COOMe r.t, 30 min, (65%). OHOH OTBS OTBS OTBS OTBS 24 h, (75%). 18 EXPECTED O O OBTAINED 21 Abstract As the above synthetic route gave the unexpected and unwanted epi compound (at the methoxy center) a small modification was applied in the retro synthetic strategy as given in Scheme 4. In this the epoxide compound 3 could be obtained from the key inter mediate 22 by extending the 10-alcohol. The key intermediate 22 could be obtained from olefin 23 which in turn could be obtained from compound 6 in a similar manner as mentioned above. Modified retro synthetic strategy. O OH OH OMe OH O OMe OMe OMe O O COOMe COOMe O OH O 3 OMe 22 6 23 . Scheme 4 The alternate strategy devised was delineated in Scheme 5. Zn mediated ring opening reaction of iodo compound 14 gave the dimethoxy olefinic alcohol 23. Dihydroxylation of the double bond followed by acetonide protection gave compound 25. This compound 25 when subjected to either protection of the hydroxyl group or deprotection of the dimethoxy ketal resulted in decomposition OH OMe Zn, EtOH. OMe O OMe OMe COOMe ref, 2 h, (87%). OH OsO4 , NMO, Acetone:Water CSA, Acetone OMe COOMe r.t, 30 min, (65%). COOMe 24 h, (65%), I OHOH 14 23 24 OH OMe OMe COOMe Decomposed subjected to any reaction O O 25 Scheme 5 5 OH OMe OMe OMe COOMe O O 25 Abstract As the above conversion was unsuccessful, we tried to synthesize the epoxide intermediate 26 which may be converted to the target molecule 1 and also can be converted to 27 which is biologically active (Fig-2). The details were outlined in Scheme 6. OH R= CH 2 Ph OR' OH R= Allyl OMe R= OT BDP S OR O O H N R'= 26 O 27 Cl O Fig-2 Reduction of the ester in 23 gave homoallylic alcohol 28. The m-CPBA epoxidation of the homo allylic alcohol 28 gave the epoxide 29. This epoxide moiety decomposed when subjected to any reaction i.e either protection of the 1o-alcohol or deprotection of the ketal. Therefore the p-alcohol in 28 was first protected as its TBDPS ether using TBDPSCl followed by epoxidation of the double bond with m-CPBA afforded the epoxide compound 31. Deprotection of the ketal to ketone 32 was carried out using CSA, acetone and water in 9:1 ratio. Chelation controlled reduction of the ketone 32 with Et3B and NaBH4 gave the syn diol 26. unfortunately the noe studies revealed that this epoxide compound is epi at the epoxide center 33. OH OH OMe OMe DIBAL-H, DCM OH OMe OMe OH TBDPS-Cl, IMD. O 29 OH OMe OTBDPS m-CPBA, DCM. O 31 OH OH OH OH Et3 B, NaBH4 . OTBDPS OTBDPS THF, 1h, (87%). O 26 EXPECTED O 33 OBTAINED Scheme 6 6 OH OMe O CSA, Acetone:H2 O OMe OTBDPS 0-r.t, (65%). 1 h, (76%). 30 28 Decomposed when subjected to any reaction. OMe OH 1 h, (78%). 28 OH OMe DCM, 1 h, (73%). OMe m-CPBA, DCM. OMe OH COOMe -780 C, 30 min, (66%). 23 OH OMe OTBDPS O 32 Abstract As the above synthetic route again gave the unexpected and unwanted epi compound (at the epoxide center) the strategy was slightly modified as given in Scheme 7. Dihydroxylation of the double bond in 28 with OsO4-NMO gave the tetrol 34. Protection of the 1,2- di hydroxyl groups and deprotection of the ketal to ketone was achieved in one pot by treating with CSA in acetone. The 1o-hydroxy group in 35 was masked as its TBS ether and chelation controlled reduction of the ketone with Et3B and NaBH4 gave the syn diol 37. Protection of the two sec-hydroxyl groups with 2,2 DMP followed by deprotection of the TBS group with TBAF in THF resulted in required cyclohexane moiety 22. The stereo chemistry of the key intermediate 22 was further confirmed by the NOESY and COSY studies. Further Studies are under progress in our lab to complete the total sythesis of fumagillin 1. OH OMe OMe OH OH OMe OsO4, NMO, Acetone:H2O OMe OH 24 h, r.t.(75%) . OH 0 0C, 3 h, (78%). OH THF, (78%). OTBS O O 37 OH DCM, 1 h O O CSA, 2,2-DMP. Acetone, r.t, (78%). O O 35 O OH Et3B, NaBH4, O O O TBS-Cl, IMD CSA, Acetone OHOH 34 28 OH O O TBAF, THF OTBS O O 38 r.t, (89%). OH O O 22 Scheme 7 7 OTBS 36 Abstract CHAPTER II, Section-B: This section describes the formal synthesis of ovalicin by carbohydrate approach. During recent years abnormal angiogenesis has been recognized as a common feature for many proliferate diseases viz. diabetic retinopathy, psoriosis, rheumatoid arthritis including cancer. Thus it has become increasingly importance to inhibit angiogenesis as one of the alternative promising method for cancer therapy. Towards these studies, ovalcin 3 isolated from cultures of Pseudorotium ovalis stock was found to be non-toxic, non-inflammatory and stable at room temperature for years bearing equal potency as AGM 1470 2 and enhanced potency compared to fumagillin 1. O Me O OMe O O O O OH Me O OMe N Cl O O AGM 1470 2 O Fumagillin 1 O Me O OH OMe O Ovalcin 3 Retro synthetic strategy: Our synthetic approach towards the ovalcin is based on chiron approach and utilizes two key steps, an olefin metathesis reaction and the zinc mediated ring-opening reaction to afford the chiral olefinic alcohol. Our retrosynthetic approach revealed a key intermediate 4 which is formed by the disconnection of the side chain and our main target was to synthesise this fragment 4 as the conversion of 4 to final target was well documented in the literature. It was envisioned that the key fragment 4 could be easily prepared by Grubbs RCM of 5, which in turn is obtained from fragment 6. The fragment 6 is easily prepared from D-ribose via a sequence of reactions (Scheme 1). 8 Abstract Retro synthetic stategy: O Me O OH OMe O 3 O O OH + OMe 4 OTES OH OMe 4 OTES OPMB D(-)ribose O Me OMe OTES 5 O I OBn OH Br O Me 6 Scheme 1 Commercially available D(-)ribose was protected as its 2,3-O-isopropylidene derivative and the primary hydroxyl group was protected as tert-butyldimethylsilyl ether with TBDMSCl and imidazole in DCM. The resulting compound 8 was homologated to compound 9 by using Corey-Chekoviskies reagent. The compound 9 was taken and protected as its MPM ether 10 with para-methoxybenzyl bromide. Desilylation of compound 10 with TBAF afforded free alcohol 11,which was further converted to iodide 6 with iodine, triphenyl phosphine and imidazole (scheme 2). HO O OH 2, 2-DMP, acetone, pTSA, r.t, 72%. O Me OH OH OH O TBDMSO TBDMS-Cl, imidazole, CH2Cl2, r.t., 98%. O TBDMSO O Me O Me 7 Me2SO=CH2, O TBDMSO O Me DMSO, 20 0C, 60%. OH NaH, PMB-Br, Et2O, 0 0C-r.t., 3 h, 97%. TBDMSO O O Me Me 10 O Me 9 TBAF, THF, 30 min. 98%. O HO O Me OPMB O Me I2,TPP, imidazole, toluene, reflux, 2 h, 80%. O I O Me 9 OPMB O Me 6 11 O Me 8 O OPMB OH Scheme 2 Abstract By applying the commonly used protocol for zinc mediated ring opening reaction compound 12 was obtained in good yields. The resulting free alcohol in 12 was masked with NaH and benzyl bromide as its benzyl ether 13. Deprotection of the acetonide group from compound 13 was achieved using pTSA to give diol 14. The diol 14 was selectively protected as the corresponding TES ether 15 at the allylic position under lower temperatures and simultaneously as methyl ether 16 with methyl iodide. The compound 16 on PMB deprotection with DDQ provided primary alcohol 17 which was oxidized to aldehyde 18 using TPAP and NMO conditions. Vinylation of the aldehyde 18 afforded allyl alcohol 5, which could be used for the metathesis reaction to get the required basic skeleton (Scheme 3). I O OBn OH OPMB Zn, EtOH OPMB O Me reflux, 2 h, 95%. O O Me Me 6 OPMB NaH, BnBr, THF, O Me 0 0C-r.t. 12 h, 85%. O Me 13 12 pTSA, MeOH, r.t. 30 min. 98%. OH OH TESCl, imidazole, OH OBn DCM, 00 C, 1 h, 84%. OMe DDQ, CH2Cl2:H2O (8:2), r.t., 2 h, 61%. 16 vinyl magnesium bromide THF, r.t., 90 min., 75%. OTESOBn NaH, MeI, THF, 0 0C-r.t., 4 h, 95%. 15 14 OTESOBn OPMB OPMB OMe OPMB O Me TPAP, NMO, CH2Cl2, OH OTESOBn 17 OMe OH OTESOBn 5 Scheme 3 10 r.t., 1 h, 80%. OMe CHO OTESOBn 18 Abstract Thus compound 5 was subjected to metathesis using Grubb’s 1st generation catalyst to afford the compound 19.The allylic alcohol 19 was converted to enone 20 with benzyl ether was achieved with Pd(OH)2 under hydrogen atmosphere to afford the compound 21. One carbon Wittig reaction on ketone with methyltriphenylphosphonium iodide yielded olefin 22. Epoxidation of the olefin with m-CPBA afforded the known key intermediate 4 (Scheme 4). OMe OH OBn CH2Cl2, r.t., 2 h, 98% OTESOBn 5 O OH (Cy3P)2Cl2Ru=CHPh 10 mol% OBn IBX, DMSO, CH2Cl2 OMe OTES 19 r.t., 1 h, 69% OMe OTES 20 O OH H2 atm. 87% Pd(OH)2, THF OMe OTES 21 O OH CH3PPh3I, KOtBu THF, 0 0C-r.t., 5 h, 87% ref OMe OTES 0 0C-r.t., 3 h, 85%. 22 O OH OMe OTES mCPBA, CH2Cl2, Me O OH OMe O 3 4 Scheme 4 This intermediate 4 was already converted to ovalicin in further few steps as reported earlier by Barton et al. Thus we have accomplished formal total synthesis of ovalicin. In conclusion, we have described an efficient formal total synthesis of ovalicin by chiron approach. Since all the reactions are high yielding, this strategy could be applied for multigram scale synthesis of the key intermediate, which can be derivatized further for the synthesis of several other analogues for various biological activities. 11 Abstract Chapter-III. Section-A: This section describes introduction -amino phosphonates, -hydroxyl amino phosphonates and previous approaches to the synthesis of -amino phosphonates. Section-B: This section describes present work on -amino phosphonates and hydroxyl amino phosphonates. The synthesis of -amino phosphonates has attracted much interest because of their biological activity and structural analogy to -amino acids. As a result, a variety of synthetic approaches have been developed for the synthesis of -amino phosphonates during the past two decades. Recently, ionic liquids have particularly gained recognition as possible environmentally safe and alternative solvent to conventional molecular organic solvents for the development of waste-free chemical processes. Ionic liquids are widely employed as novel and recyclable reaction media for a variety of transformation including asymmetric hydrogenation reaction, oxidation, Friedel-Crafts alkylation and acylation, Diels-Alder reaction, esterfication reaction, Heck-reaction, Baylis-Hillman reaction, Wittig reaction, aromatic nitration, Baker’s yeast reduction, lipase induced transetserfication, acetylation reaction, alkene metathesis, asymmetric epoxidation, Oalkylation, hydroformylation, Fries rearrangement, allylation reaction, Pechmann reaction, Knoevenagel condensation, single-electron transfer reaction, and polymerization reaction, etc. N N BF4- N Liquid (Hydrophilic) N PF6- Liquid (Hydrophobic) bmimPF6- bmimBF4- bmimBF4- : 1-Butyl -3-methyl imidazolium tetraflouroborate. bmimPF6- : 1-Butyl -3-methyl imidazolium hexaflourophosphate. 12 Abstract Herein, the ionic liquids (bmimBF4 & bmimPF6) have been proved as an efficient catalyst and reaction medium for the three component coupling of a carbonyl compound, amine and diethyl phosphite to yield various -amino phosphonates (Scheme 1). This method describes a general procedure for producing biologically important -amino phosphonates. R-CHO + R'-NH2 + bmimBF4 HOP(OEt)2 or bmimPF6 r.t HN R' OEt R P O OEt Scheme 1 We first investigated the reaction between a simple, commercially available benzaldehyde 1a, amine 2a and diethyl phosphite 3 by stirring equimolar quantities of the three components in ionic liquids (ILs) for an appropriate time (Table-1) and isolated the desired -amino phosphonate 4a in excellent yields (Table 1) (Scheme 2). CHO NH2 NH + (HO)P(OEt) + r.t., 5.0 h, 90% 2 1a ILs 2a 3 P(O)(OEt)2 4a Scheme 2 Similarly several aldimines (generated in situ from aldehydes and amines) reacted with diethyl phosphite under similar reaction conditions to afford the corresponding amino phosphonates in high yields. In all cases, the reaction proceeded smoothly at ambient temperatures with high selectivity (Table 1). 13 Abstract Table-1 Aldehyde Entry NH2 CHO a bmimBF4 Amine CHO NH2 b OH bmimPF6 Time/h Yieldb% Time/h Yieldb% 5.0 90 8.0 84 7.5 88 95 9.5 85 6.5 85 09 9.0 81 5.0 91 8.0 87 5.0 90 8.0 85 6.5 90 6.0 Cl CHO NH2 CHO NH2 c 3.5 5.0 5.5 5.0 4.5 7.0 5.5 4.5 MeO d F NH2 e S CHO CHO NH2 f g () 4 NH2 CHO CHO h 9.5 70 13.0 68 11.5 70 NH2 8.0 a 75 Products were characterized by 1H NMR, IR and mass spectroscopy b Isolated yields of products 14 Abstract Both aromatic and aliphatic aldimines produced excellent yields of products. However, the reactions of aliphatic aldimines took longer reaction time compared to aromatic imines because of the low reactivity of aliphatic aldehydes than aromatic. The reactions are clean and complete within 5-12 hours. The reaction conditions are very mild and amino phosphonates are exclusively formed without the formation of any undesired side products. Encouraged by the above results, we tried to apply the same ILs system for the synthesis of α-hydroxyl amino phosponates. Although a large number of methods is available for the synthesis of -amino phosphonates, only few methods are reported for -hydroxylamino phosphonates. Further more there are no examples on the use of ILs as promoters for the three component coupling of aldehydes, hydroxylamines and diethyl phosphite to produce -hydroxylamino phosphonates . The treatment of benzaldehyde and phenyl hydroxylamine with diethyl phosphite in ILs afforded the corresponding -hydroxylamino phosphonate in excellent yield (Scheme 3). HO Ph-CHO + Ph-NH-OH + HOP(OEt)2 bmimBF4 or bmimPF6 r.t N Ph OEt Ph P O OEt Scheme 3 In a similar fashion, various aldehydes and aryl hydroxylamines reacted smoothly with diethyl phosphite to give the corresponding -hydroxylamino phosphonates in excellent yields (Table 2). The reactions proceeded efficiently at ambient temperature with high selectivity. No trace amounts of -hydroxy phosphonates are obtained as a result of the reaction between the aldehyde and diethyl phosphite. However in the absence of ILs the reaction did not yield any product even after a long reaction time. 15 Abstract Table-2 Aldehyde Entry bmimBF4 Amine Time/h a CHO CHO b H N OH H N OH bmimpF6 Yieldb% Time/h Yieldb% 3.5 89 2.5 92 3.5 90 4.0 87 4.5 90 5.0 85 2.5 92 92 3.5 87 3.0 89 4.5 85 3.5 92 4.5 90 3.0 95 4.0 89 MeO MeO CHO H N c OH MeO OMe CHO H N d CHO e H N H N f S O OH OH CHO H N g OH OH CHO a Products were characterized by 1H NMR, IR and mass spectroscopy b Isolated yields of products 16 Abstract In addition to its simplicity and milder reaction conditions, this method is even effective with aliphatic and unsaturated aldehydes which normally produce low yields. The present method does not require any additives or promoters to proceed the reaction. Another important feature of this reaction is the survival of a variety of functional groups such as olefins, ethers, and halides under the reaction conditions. The advantage of the use of ILs as novel reaction media for these two transformations is that these ILs were easily recovered and reused. In summary, we have demonstrated a novel and efficient protocol for the synthesis of -amino phosphonates and -hydroxylamino phosphonates which can serve as peptide mimetics by using ILs as efficient catalysts and reaction medium. The method is effective for aromatic, aliphatic, unsaturated aldehydes and provides excellent yields of the products, which makes it useful and attractive process for the synthesis of α-amino phosphonates and -hydroxylamino phosphonates. It is believed that this method presents a better and more practical alternative to the existing methodologies for the synthesis of -amino phosphonates and -hydroxylamino phosphonates. 17