Week 8-Bioinformatics/Proteomics
advertisement

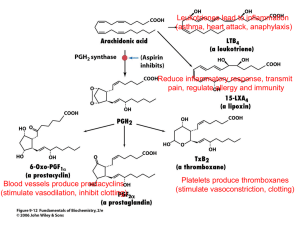
Laboratory 4-Bioinformatics/Proteomics (Based on laboratory procedures written by April Bednarski) Introduction: The basic unit in biology is the cell. Therefore, all organisms on earth are made of cells. Some organisms are unicellular, whereas others are multi-cellular. In biology cells are characterized into two basic categories, prokaryotes and eukaryotes. Prokaryotes include unicellular organisms such as bacteria, which do not contain membrane bound organelles. Eukaryotes include both unicellular organisms (such as some fungi) and multi-cellular organisms, which do have membrane bound organelles. Proteins carry out almost all of the important functions of the cell, such that it can survive. For instance, proteins are important for such processes and functions as replicating DNA, breaking down glucose to harvest energy (cellular respiration), providing structure to the cell, sensing the environment around the cell, responding to a variety of stresses, as well as a host of other functions that are too numerous to name. Many proteins between cells are conserved. In this lab we will use various computer based tools to study protein sequences, and conservation of proteins between species. The first set of proteins we will work with is the COX-1 and COX-2 proteins. In the second part of the laboratory, you will be given an unknown protein sequence. Learning Objectives: 1. 2. 3. 4. Learn how to use NCBI blast software Learn how to analyze protein sequences using ClustalW Learn how to study a protein sequence and find homologs in other species Learn to determine the level of conservation of amino acid sequence between homologous proteins 5. Learn to find an amino acid substitution in a protein sequence 6. Learn to find information about the function of a protein 7. Learn that experiments in Cell Biology today are sometimes done in silico (on the computer) Experimental Objectives: 1. To determine the function of both COX-1 and COX-2 proteins 2. To find homologs of COX-1 and COX-2 and determine the level of conservation between homologs 3. Determine the identity of a given unknown protein sequence 4. Determine whether the unknown protein sequence has an amino acid substitution 5. Use the actual sequence of your unknown protein you found to find homologs Background: In the laboratory, we will work through an experiment using web-based bioinformatics programs to study protein sequences. Almost all of our cellular functions are carried out by proteins. Many are enzymes, but other proteins have other functions. A protein is made by polymerizing (stringing together) amino acids. Each type of protein has its own unique amino acid sequence. In eukaryotes, there are 20 amino acids. Many of these amino acids are non-polar. However some are polar, and others are charged either positively or negatively. In the protein sequences we will study in this lab, the amino acids will be noted by a single letter designation. The key for the designations is in the figure on the right. You can find the polarity information for each amino acid in your Cell Biology book. In this laboratory, we will start with a protein of interest, in which we know its amino acid sequence, and search to determine whether the protein has any homologs. Homologs are proteins from different species that have similar sequence and function, which have been conserved. When we talk about conservation, we are stating that both the protein sequence and function have been maintained through evolution. When we study protein sequences, we mean that the amino acids at a given position in the protein sequences we are comparing are identical. Homologs are protein sequence The first part of the experiment we will study the protein sequence for the enzymes COX-1 and COX-2 (which also goes by the name PTGS2). The bioinformatics programs we will use are Protein, BLAST, RefSeq, PubMed and ClustalW. Protein is a database of protein sequences curated by NCBI, RefSeq is a database of sequences that is edited by NCBI, and is non-redundant. This means that NCBI has determined the strongest sequence data for each gene. BLAST is a program by which we can take a specific protein sequence and search for protein sequences that are conserved (evolutionarily related). PubMed is a database that is curated by NCBI, and contains information about proteins, as well as protein sequences. Lastly, ClustalW is a program that allows you to enter a series of protein sequences that you believe are similar, and further compare them. COX-2 (PTGS2)is called prostaglandin H2 synthase-2 and cyclooxygenase-2 (COX-2). COX-2 has been thoroughly studied because of its role in prostaglandin synthesis. Prostaglandins have a wide range of roles in our body from aiding in digestion to propagating pain and inflammation. Aspirin is a general inhibitor of prostaglandin synthesis and therefore, helps reduce pain. However, aspirin also inhibits the synthesis of prostaglandins that aid in digestion. Therefore, aspirin is a poor choice for pain and inflammation management for those with ulcers or other digestion problems. Recent advances in targeting specific prostaglandin-synthesizing enzymes have lead to the development of Celebrex, which is marketed as an arthritis therapy. Celebrex is a potent and specific inhibitor of COX-2. Celebrex is considered specific because it doesn’t inhibit COX-1, which is involved in synthesizing prostaglandins that aid in digestion. This is a remarkable accomplishment given the great similarity between COX-1 and COX-2. This achievement has paved the way for developing new therapies that bind more specifically to their target and therefore have fewer side effects. Understanding the enzyme structures of COX-1 and COX-2 helped researchers develop a drug that would only bind and inhibit COX-2. Many of the types of information and tools used by researchers for these types of studies are freely available on the web. In this tutorial, and throughout this lab course, you will be introduced to the databases and freely available software programs that are commonly used by professionals in research and medicine to study genes, proteins, protein structure and function, and genetic disease. Experimental Procedure: PART A: Follow these directions to access the entries for PTGS1 (COX-1) and PTGS2 (COX-2) by using the NCBI Website: 1. First, go to the NCBI homepage by going to: http://www.ncbi.nlm.nih.gov 2. To access the protein database find the word “Search.” From the database pulldown menu select “Protein.”. Type “PTGS” in the search box, then click “Go.” 3. Scan the results for the “Homo sapiens” entries. There should be one called “PTGS1” and one called “PTGS2.” We do not want the references to the enzyme found in the yeast Schizosaccharomyces pombe 972h. 4. Select each entry by clicking on its name, then read the paragraph under the “Summary” section for each entry. After reading the “Summary” section for both of these proteins, answer the questions below. 1. PTGS1 and PTGS2 are isozymes. Isozymes catalyze the same reaction, but are coded by separate genes. What types of reactions to PTGS enzymes catalyze? Also, what pathway are these enzymes a part of? 2. How is the expression of PTGS1 and PTGS2 different? 3. Which protein (COX-1 or COX-2) would you want to inhibit to stop inflammation? The next two questions are not discussed in the summaries- just read the questions and think about the answers. 4. The drug Celebrex selectively inhibits PTGS2 while aspirin and other NSAID’s inhibit both PTGS1 and PTGS2 in the same way. Why do you think researchers wanted to discover a selective inhibitor to PTGS2? 5. Copy the protein sequences for COX-1 and COX-2 into Microsoft Word, and properly label them. Attach your sequence information to the back of your worksheet for this laboratory. PART B. Now let’s go and search for homologs of both COX-1 (PTGS1) and COX-2 (PTGS2). To do this we will use the program BLAST. 1. From your work with COX-1 above, go and find the protein sequence in your Microsoft Word file, and copy it. 2. Go to NCBI blast by typing the link http://www.ncbi.nlm.nih.gov/BLAST/ 3. Go to the menu below and select protein blast 4. Paste your sequence in the sequence box at the top of the webpage, and then click the BLAST button. (Note: It may take some time as the program searches the database for homologs) Paste sequence here 5. When the search is complete, you will be taken to a screen that contains the list of homologs. Next to each homolog will be the latin name of the species from which the homolog has come from. In the list, next to the names, there is an e value. In general, the smaller the e value, the greater the chance that the identified sequence is a homolog. Identified sequences with an e value of less than 10-5 are usually classified as homologs. 6. Below the list, you will see the sequence you blasted (query) compared to the sequence of the homolog (subject). Above the sequence comparison, there will be some information. Listed in that information, is the name of the subject protein, and the species from which it came. Also listed in the information is the % identity and % similarity. The % identity shows the percentage of amino acids that are identical at the same positions between the two protein sequences being compared. The % similarity shows the percentage of amino acids that have similar characteristics at the same positions between the two protein sequences being compared. 7. Go through the same process using COX-1 as your query sequence. Questions For Part B: 1. After blasting the COX-2 sequence, how many potential homologs came up on the list. How many of these homologs have an e-value of less than 10-5. 2. List the five closest homologs for COX-2. Also, next to each homolog, place the name of the species it came from. 3. For each of the five closest homologs, determine both the identities and similarities and write them in the space below. 4. Why do you think COX-1 came up in the blast search? Please relate that to the information about function that you found in the previous section. 5. Given the level of sequence specificity, would you expect aspirin to have an effect on both COX-2 and COX-1? Why do you think this is the case? Part C: Sequence Analysis Using ClustalW Now let’s further analyze the COX-2 protein sequence as compared to its five closest homologs. To do this, we need to get the sequences for these homologs and copy them into Microsoft Word. To do this, go to the information listed above each sequence comparison. 1. Click on the link with the protein name. This will bring you to a page which has the protein sequence of the subject (one of our homologs) at the bottom. 2. The sequence is unfortunately not in a proper format to use in ClustalW (the program we will next use). The format the sequence must be in is FASTA. Go to the top of the page, where you see Display. In the box next to it, it should say GenPept. Click on the box, and change the format to FASTA. Change the format using this box 3. The sequence is now displayed in FASTA format. The FASTA format has a title line for each sequence that begins with a > followed by a description of the sequence. On the line below the description will be the sequence itself. Copy all of this information, and paste it into Microsoft Word. Above the sequence, place a label, such that we will know which sequence it is. Sequence Information Protein Sequence 5. Do this for the 4 next closest homologs. You can paste them into the same Microsoft Word File. 6. Now let’s go to ClustalW. ClustalW can be found at http://www.ebi.ac.uk/Tools/clustalw/index.html. 7. Let’s paste all of our FASTA sequences into the window at the bottom of the page. Be sure to include all of the FASTA information, but leave out your sequence label. Also be sure there are no line breaks in any of the protein sequences. Leave a space between each sequence you paste in the box. 8. Once all of the sequences are successfully pasted in the box, press run. At this point ClustalW will try to align your sequences. Once our sequences are aligned, the identities will be made clear by the * symbol below the amino acid. Similarities will be denoted by either a single dot (.) or a double dot (:). Sequence alignment 9. Once the alignment comes up, click on view alignment file. This will bring us to a page that just has the alignment. Save this alignment. You will need to print this out to turn in with your worksheet. 10. Go back to the initial alignment page, and view your sequence score. View the output- the SCORES table: SeqA Name Len(aa) SeqB Name Len(aa) Score =================================================== 1 dog 604 2 cow 604 90 1 dog 604 3 mouse 604 89 Note that different specific combinations are examined; DOG TO COW for example. You would expect a higher SCORE (right column; similarity of the gene sequence) between a human and a chimpanzee (both primates), than say between a human sequence, and a frog sequence. What are the similarity scores for the same sequence when comparted to COX-2? Sequence 1 _________ Score ________ Sequence 2 _________ Score ________ Sequence 3 _________ Score ________ Sequence 4 _________ Score ________ Sequence 5 _________ Score ________ 11. Go to the bottom of the page; view the Cladogram. Go to this source to learn more about cladograms: http://en.wikipedia.org/wiki/Cladogram. Summarize your findings about the evolution of this enzyme in your lab notebook. Questions Part C: 1. Print out the FASTA and the ClustalW sequence comparison files, and affix them to this worksheet. 2. For each of the five sequences, list the comparison scores to human COX-2 in the space provided below. 3. Which sequence had the highest comparison score to human COX-2, and which had the lowest comparison score to human COX-2. Does this match with how closely related you think the species are (i.e. mammal vs. mammal as compared to say mammal vs. amphibian)? 4. How does this scoring list compare with the scoring list from BLAST? 5. Make a copy of your cladogram, and affix it to the worksheet below. From your work, describe how the sequences fall out in your cladogram.