CRIS and Research Documentation at ADHB
advertisement

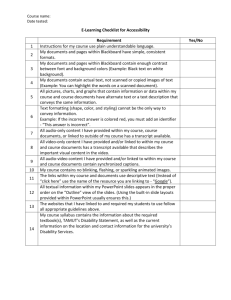
CRIS and Research Documentation at ADHB 1. Introduction Clinical records at ADHB are maintained via the Clinical Record Information System (CRIS). This system involves the digital scanning all clinical records and documents, which can then be securely viewed by authorised users via a PC at any geographic location across the ADHB network. Authorised staff can also download records to a laptop computer at off-site locations. Clinical records are scanned in full colour (i.e. not black and white). This ensures a high quality image that matches the original content in the paper record. Once a document is scanned it cannot be altered in any way*. The digitised clinical record is stored on a magnetic disc (the primary storage system) and backed up to an optical disc within seconds of being committed to the primary system. All systems meet the requirements of FDA 21 CFR Part 11. The information stored on CRIS will be transferred from magnetic/optical storage to emerging media at appropriate time intervals, as technology advances. ADHB uses a ‘scan on demand’ model, whereby existing records are scanned in their entirety prior to a patient’s booked inpatient or outpatient visit. Old records that are not reactivated by a new patient visit are not scanned. Records that have not been scanned will continue to be available in paper form. 2. Access to CRIS Where appropriate, ADHB staff are issued with a CRIS User ID and passwords to facilitate on-line access to required records. Access to hard copy records is available via the standard record request procedures (i.e. by contacting the Clinical Record Department on site). External study monitors are permitted access to specific records for specified periods. To arrange this, contact the Clinical Records Department on ext. 7274. For all scanned records, the Clinical Record Department will arrange for the external monitor to be issued with a CRIS logon/User ID and password. It is ADHB policy not to share your password. External study monitors are welcome to use the viewing room facility at the Grafton site. This facility is equipped with PCs to provide access to CRIS. To book these rooms, contact the Clinical Record Departments at Grafton on ext. 7274. * Only authorised clinical record department staff with an appropriate security level can delete documents in CRIS. All transactions within the system (document viewing, printing, deletion, etc.) are recorded in the system’s Audit Log. CRIS training and support material is available from the Clinical Records site on the ADHB intranet: <http://ahsl85_gl/ClinicalRecords/Cris_Training/Training.htm> Research Office June 2006 Page 1 of 4 CRIS and Research Documentation at ADHB Personal support is also provided via the Clinical Workstation Training & Support Unit (CWTSU) Helpdesk on ext 27000. Should research sponsors require further information regarding the storage media, how the data is backed up and the processes for managing document deletion/amendment, they should be referred to Linda Fletcher, Manager, Patient Information Services, ext. 24074. 3. Clinical Trial Alerts If there is a need to alert others of a patient’s participation in a clinical trial, then the trial Principal Investigator, or their authorised representative, should complete a Clinical Alert Notification Form (document order number CR0008) for each study participant. This form should include: the ticked “Clinical Trial” box the Short Project Title (as per Research Consent Document or Information Sheet) the Ethics Committee (or ADHB) study number a contact name and phone number the NHI number The study information sheet Completed Clinical Alert Notification Forms are sent to the CRD, Building 21, Grafton. The Alert is added in CMS – CMS informs CRIS that there is an alert on this patient, but the details of the Alert do not go across to CRIS. CRIS displays a message to the end-user ‘A Clinical Alert exists for this patient. Please check documentation in the record / CRIS for details’. The Alert Notification Form is scanned and can be viewed under the Alerts Tab (The Alert Notification Form is processed by CRD staff and then comes to the Scan Centre for urgent processing). 4. Clinical trial documentation All research related documentation is to be sent to the Scan Centre via the blue back pocket, used for clinic documentation. The patient’s NHI number should be on every page. All documentation will be scanned within 24 hours of receipt by the Scan Centre. Indexing All research documentation will be indexed to categories in the mainstream record, i.e. clinical notes to the clinical notes tab, laboratory results to the result type, etc. The exception to this will be anything other than clinic notes, lab results, or investigations or information and consent forms, e.g. trial worksheets and other miscellaneous research documents. These documents should be stamped with a clinic trial documentation stamp. Research Office June 2006 Page 2 of 4 CRIS and Research Documentation at ADHB Note: This is the responsibility of the research area to ensure these documents are stamped. Failure to do so may result in errors in indexing if these documents are not recognised as clinical trial documentation. Clinical Notes Research units have the option of developing and using customised barcoded clinical note forms. How to arrange these forms can be discussed with Sue Guthrie, Health Information Manager, on ext. 4050. Consent Forms & Information Sheets All Consent Forms for research participants must be accompanied by the current approved version of the Information Sheet. The Information Sheet and Consent Forms are considered a single legal document. These will be indexed to Consent Form - Clinical Trial and viewable under the Consent tab. Laboratory Results Researchers should not send hardcopy ADHB laboratory results for scanning as all ADHB laboratory results are sent electronically into CRIS. Research areas should use the clinical notes to document that they have seen a result and any action that was taken. 5. Corrections to scanned clinical trial documents The process for correcting documents that have been scanned is to print the scanned document, make the changes in the standard method (cross-out, correct, sign, date), then send the revised paper back for scanning. The revised documentation should be addressed to the CRIS Business Administrator, Clinical Record Department, Building 21, Grafton. It is important that the revised documentation is accompanied by a special note requesting that the original document be deleted and replaced with the revised documentation. 6. Notification to Sponsors of the move to electronic based records In November and December 2005 letters were sent out to all the known sponsors of research (current and past) informing them of the transfer to electronic based records and that overtime the paper copies would be destroyed. They were given 8 weeks to respond with any queries or requests. No responses have been received. Of note, Clause 9 of the Health (Retention of Health Information) Regulations 1996 provides that there is no requirement to keep information in any particular form. Information can be retained in what ever form the health provider deems fit. It is therefore permissible to hold records in paper then scanned form. Once they have been scanned, these scanned records can be deemed the official record and the paper copies destroyed. Research Office June 2006 Page 3 of 4 CRIS and Research Documentation at ADHB 7. References Regulations and guidelines (excerpt from the GCP Journal, April 2003): a. Retention times are referred to in the Note for Guideline on GCP (CPMP/ICH/135/95). Section 5.5.11 notes that sponsor-specific essential documents should be kept for at least two years after the last marketing application approval in an ICH region and until there are no pending or contemplated marketing applications there, or at least two years have elapsed since formal discontinuation of an investigational drug’s clinical development. Section 5.5.12 says sponsors must notify investigators and institutions in writing when trial-related documents are no longer needed. b. EU storage requirements are laid out in the draft Detailed Guidelines on the Trial Master File and Archiving, which was produced to supplement the EU clinical trial directive (2001/20/EC). It details the minimum set of documents to be retained, the quality of the documents to be archived, minimum standards for storage conditions, media transfer and certified copies and retention times. c. US requirements are specified in FDA regulation 21 CFR Part 11. This provides criteria under which the FDA accepts electronic records, electronic signatures, and handwritten signatures executed on electronic records as being equivalent to paper records with handwritten signatures. d. Health Information Management Service at Stanford University Medical Center. http://www.clinicaltrials.stanford.edu/med_records.pdf e. Medical University of South Carolina stating that scanned records may be destroyed after a period of 6 months: http://www.musc.edu/uma/compliance/policies/C001.pdf Research Office June 2006 Page 4 of 4


![Appointments: Manual Booking using [ALT-M] in conjunction](http://s3.studylib.net/store/data/007588400_2-a89991296ab31df74067d7b72cd8b787-300x300.png)