emi12658-sup-0001-si

Supplementary data:
Pyrosequencing: Barcoded 454-pyrosequencing followed by quality filtering yielded in an average sequence length of about 300 nt. After exclusion of reads assigned to chloroplasts, mitochondria and the cyanobacterial genus Nostoc , which is harbored in internal cephalodia of the thalli, two pyrosequencing data sets at a genetic distance of 3% were generated. The first one was used for the comparison of the lichen thalli with its propagules and was made up of 10 samples (5 thallus- and 5 propagule-samples) collected in Johnsbach. It comprised
74,180 sequences (min: 4,891 seq., max: 10,512 seq.) representing 3,273 operational taxonomic units (OTUs). The second one was used for comparison of lichen thalli within sampling sites and was made up of 15 thallus samples (5 samples for each sampling site). It comprised 83,080 sequences (min: 3,635 seq.; max: 8,021 seq.) representing 3,972 OTUs.
Due to the difference in sequence counts among the samples both datasets were normalized to the minimal sequence count in each dataset resulting in 41,890 sequences representing 2,439
OTUs for dataset 1 and a total number of 3,117 OTUs with 54,525 sequences for data set 2.
None of the rarefaction curves were saturated, resulting in a coverage of ~51-53%.
Statistics : A non-parametric analysis of variance and a RDA based variation partitioning were used to survey the extent to which the differences in community composition can be explained - by geographic factors and by the tissue origin. Both analyses were carried out in R
(http://www.R-project.org) using adonis and varpart as implemented in package vegan
(http://vegan.r-forge.r-project.org).
Both analyses are similar, despite of their conceptual differences. Adonis uses the localities as categorical variables, while for variation partitioning a geographic distance matrix was used to capture geographic relatedness. Both analyses were run twice using different response matrices: a) the OTU community matrix obtained in QIIME, standardized using “total” method in vegan function decostand and a pairwise dissimilarity matrix obtained by analyzing the SSCP band pattern with GelCompare II 6.5 created by Applied Maths NV
(http://www.applied-maths.com/).
The results of all four analyses are highly coherent and are summarized in table S3. Only a small fraction of the variability in the datasets is explained by the analyzed factors, ranging between 30% and 41% in all analyses. This apparent lack of explanative power is coherent with the methodological constraints in amplicon studies like having identified thousands of
OTUs in a small number of samples resulting in a high variability between samples. Across
1
all analyses, most of the explained variation in community composition was due to the tissue of origin (e.g. 24-25% in adonis).
However, when the co-structuring between tissue of origin and distance was partialled out using variation partitioning in the Qiime dataset, this percentage dropped to ca. 14%. Also, an additional 5% to 9% of the variability in the studied datasets was explained by the differences between localities or by geographic distance across all analyses. However, the strong costructuring between tissue of origin and locality found in the QIIME dataset might reflect the lack of complete nestedness of this dataset. Meanwhile, when using the fully nested SSCP band dataset, geographic distances became more explanative than the tissue of origin, explaining up to 24% of the variability.
2
Figures
:
S1: Microscopic pictures of the vegetative propagules of lung lichen. a) young stage of propagules (soredia) made up of algal cell clusters wrapped by fungal hyphae (wet state). b) late developmental stage of propagules as cylindrical outgrowths stratified with fungal cortex
(isidioid soredia; dry state).
3
S2: Single strand conformation polymorphism analysis of the lung lichen thalli and vegetative propagules of the three sampling sites (Tamischbachgraben, St. Oswald/Eibiswald,
Johnsbach, white bars). M: marker; red bar: vegetative propagules; blue bar: thalli
4
S3: A) Distribution of the relative abundances of either sequences or OTUs across lichen parts. B) Venn diagram of OTUs from lichen thalli and propagules (sampled at Johnsbach).
Thalli and propagules shared 689 OTUs, but had also unique OTUs for each lichen part.
5
S4: Composition of the core microbial community of the two lichen parts at phylum, class, order and family level (starting from the inner circle). Within the Proteobacteria,
Deltaproteobacteria was the predominant group (52%).
6
S5: Principal coordinate analysis (PCoA) of bacterial communities based on unweighted, jackknifed unifrac distance matrix. A) Beta diversity of the bacterial communities of lichen thalli and propagules sampled at Johnsbach (Dataset I; 5 thallus and 5 propagule samples).
Samples were represented by: blue rectangles – lichen thalli, red points – isidioid soredia; p<
0.022 (Anosim) B) Beta diversity of the bacterial communities of the three different sampling sites (Dataset II; 5 thallus samples at each site); p< 0.001 (Anosim). Samples were represented by: green rectangles - St. Oswald/Eibiswald, blue points - Johnsbach and brown triangles - Tamischbachgraben.
7
S6: Alphaproteobacterial families on isidiod soredia. The most dominant one was
Sphingomonadaceae comprising more than 60%. Alphaproteobacterial sequences, which could not be assigned to a certain family taxon are summarized in the category “Other”. All further families found comprised only 8%.
8
Tables
:
Table S1: Alpha diversity* and richness indices of lichen thalli and vegetative propagules.
MID1
MID2
MID3
MID4
MID5
MID6
MID7
MID8
MID10
MID11
MID13
MID14
MID15
MID16
MID17
MID18
MID19
MID40
MID21
Shannon
7.1
6.17
6.71
6.83
5
7.08
6.76
6.98
7.6
7.21
7.28
7.65
7.54
7.1
7.33
5.98
7.07
6.05
5.59
MID22 4.97 0.92
*normalized to 3635 sequences/sample; 3% genetic distance.
Simpson
0.97
0.93
0.96
0.97
0.87
0.97
0.97
0.97
0.99
0.98
0.98
0.99
0.98
0.97
0.98
0.95
0.97
0.96
0.94 observed species
595
484
570
563
369
570
531
580
672
567
634
702
658
647
606
472
616
419
327
275 chao1
1173
951
1131
1122
722
1118
1065
1194
1444
1188
1268
1490
1367
1229
1249
975
1207
799
672
594
9
Table S2: Distribution of alphaproteobacterial families across all three sampling sites order
Alphaproteobacteria other
BD7-3
Caulobacterales family other
St. Oswald
1.65
Johnsbach
2.58
Tamischbachgraben
0.93
Ellin329
Rhizobiales
Rhodospirillales
Rickettsiales
Sphingomonadales other other
Caulobacteraceae other other
Aurantimonadaceae
Beijerinckiaceae
Bradyrhizobiaceae
Hyphomicrobiaceae
Methylobacteriaceae
Methylocystaceae
Phyllobacteriaceae
Rhizobiaceae
Xanthobacteraceae other
Acetobacteraceae
Rhodospirillaceae other
Rickettsiaceae other
Sphingomonadaceae
0.06
0.84
0.17
0.09
0.28
0.02
0.66
7.21
0.18
0.26
5.04
0.07
39.47
0.02
0.02
0.62
0.17
0.07
0
0.11
42.99
100.00%
0.08
1.16
0.06
0
0.38
0
0.30
8.41
0.85
0.21
3.61
0.49
34.71
0.02
0
1.04
0.15
0.36
0.08
0
45.51
100.00%
0.10
2.51
0.07
0.01
0.21
0.01
0.77
4.96
0.17
0.23
4.39
0.10
27.57
0.07
0.03
0.53
0.04
0.03
0.04
0.14
57.07
100.00%
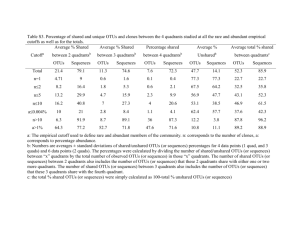
Table S3: Multivariate analysis. Statistical significance was tested with Adonis and ANOVA on the testable RDA fractions. Support values are indicated by an asterisk (*) when p<0.05 and by two (**) when p<0.01
SSCP band patterns
Adonis [R 2 ] Variation partitioning
[Adjusted R 2 ]
Qiime data (OTUs)
Adonis [R 2 ] Variation partitioning
[Adjusted R 2 ] lichen parts 0.24761 ** 0.17163 ** 0.24142 ** 0.13545 ** locality/ geographic distance 0.04580 0.23556 ** 0.08653 * 0.09173 * combination of both unexplained variation
0.07204 *
0.63455
-0.00098
0.59379
0
0.67205
0.07405
0.69877
10