Transcript
advertisement

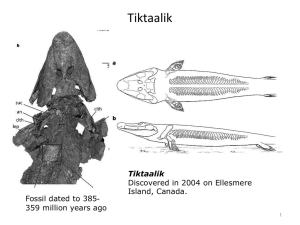
CLASS: FUNDAMENTALS I Scribe: Daniel McCullough 8-15-2011 11:00-12:00 Proof: Joe Vaughn Ryan Hemoglobinopathies & Thalassemia Page 1 of 7 I. Intro Slide [S1] II. Introduction [S2] a. Class of disorders where you have chain imbalance. i. Need equal amount of alpha and beta to make dimers and tetramers 1. Not enough alpha is alpha thalassemia. 2. Not enough beta is beta thalassemia. III. Red Blood Cells Contain Hemoglobin [S3] a. Here is a red blood cell with hemoglobin. i. The two diseases we are going to talk about are sickle cell anemia and Cooley’s Anemia. 1. Sickle cell anemia is caused by a single nucleotide change in the DNA, which causes a single AA change, which, in turn, causes all the disease consequences. 2. Cooley’s Anemia is the most severe form of beta thalassemia. a. It results when there are no beta globin chains. If all you have is alpha you can’t form alpha, beta dimers and tetramers. i. These excess alpha chains lead to the early destruction of the cell (the erythroblasts in the bone marrow). IV. Globin Gene Regulation [S4] a. Globin genes are regulated in four important ways. b. They are expressed in very high levels, specifically in erythroid cells. c. Individual globins are expressed through development. i. We make an embryonic globin in early embryos, a fetal form through most of our fetal life, and then our adult form. 1. This is called Hb switching. 2. The Hb’s that are expressed and synthesized into protein change through development. a. There is a physiological significance to Hb switching. i. Making the fetal Hb during fetal life allows it to pull oxygen from the maternal circulation…from the maternal adult Hb to the fetal Hb so the fetus can deliver it to their tissues. d. Alpha and beta-like globin genes are coordinately expressed. The red blood cell tries to make equal amounts of these things so you can get balanced expression and have plenty of tetrameric Hb. V. Human alpha and beta Globin Loci [S5] a. Both the alpha and beta globin genes are found on two different chromosomes, but each of the individual family members are found in a locus. i. For the alpha locus there is an embryonic gene called zeta and two adult genes called alpha two and alpha one. ii. Similarly, at the beta globin locus on chromosome 11, there is an embryonic epsilon gene, two fetal gamma globin genes (G gamma and A gamma), and there is two adult genes (a minor adult form called delta and the major adult form called beta). 1. In our blood streams, we have about 97% α2β2. That is Hb A. 2. We have about 2.5% Hb A2, which is composed of two alpha chains and two delta chains. 3. We have about 0.5% fetal Hb, which is α2γ2. iii. In adults, we only make adult alpha globin chains and we make mostly this adult beta, a little bit of the minor adult delta, and a half a percent of fetal Hb. VI. Erythroid Development [S6] a. All RBC’s come from hematopoietic stem cells in the bone marrow, but the source of these hematopoietic cells come from different sites during development. VII. Human Hemoglobin Switching During Development [S7] a. Very early in gestation, the first blood cells arise in the yolk sac blood islands. i. There are hematopoietic stem cells in the yolk sac blood islands that make primitive RBC’s, which make zeta and epsilon or alpha and epsilon. These are the embryonic globins found in the early embryo. ii. Then when these hematopoietic stem cells seed the fetal liver, which causes hematopoiesis to occur in the fetal liver during most of the fetal life, you produce fetal Hb. 1. You are still making the adult alpha chains, but now you begin to make this fetal beta chain called gamma. So α2γ2 is Hb F, fetal Hb. iii. Then after birth, hematopoiesis takes place in the bone marrow. CLASS: FUNDAMENTALS I 8-15-2011 11:00-12:00 Ryan Scribe: Daniel McCullough Proof: Joe Vaughn Hemoglobinopathies & Thalassemia Page 2 of 7 1. The bone marrow hematopoietic cells produce RBC’s that predominantly make the adult globin chains. a. The major one, Hb A. The minor one, Hb A2. VIII. Beta Globin Gene Switching [S8] a. This is a schematic showing how our beta-like globins switch during development. i. The Y-axis is percent of total beta chains, and the X-axis is time. The vertical line shows birth. ii. In the early embryo, we make the embryonic epsilon chains. They decrease and then through much of fetal life we make the two fetal genes. 1. Then at birth, we have about 70-80% fetal Hb and about 20-30% beta globin. a. This is important b/c if you are a patient that has sickle cell disease or Cooley’s anemia, where you don’t have any beta chains, at birth you are okay b/c you have high levels of fetal Hb. i. You can survive on fetal Hb in your RBC’s until your RBC’s stop expressing the fetal genes. ii. When they make that final switch from fetal to adult beta, if you are missing the beta globin gene then you have problems. 1. This occurs after birth, and about the first year to year and a half after birth, our fetal Hb levels fall from 70-80% down to 0.5%. iii. We wanted to model these disorders in the mouse, but we had a big problem. 1. The problem is that mice don’t have a fetal Hb. They have two embryonic globins, called beta h1 and epsilon Y. 2. The adult beta major and beta minor, as they are called in the mouse, they come on early in fetal life. a. About 2/3 of the way through gestation, they basically have 100% of their adult globin. 3. If you wanted to try and make a mouse model of beta thalassemia, you can just delete these two genes and make a mouse that is heterozygous for this deletion. a. These heterozygous mice will have about half of the amount of the beta globin in their RBC’s and they get beta thalassemia. b. However, if you breed two heterozygous mice together, then ¼ of their offspring would have a homozygous deletion. They wouldn’t have any adult beta, and the mouse would die in development. i. So, we had to figure out a way to give the mouse fetal Hb, and we humanize these mice by giving them human globins. 1. We take human DNA, put them into mouse embryos, and we can get mice that survive on 100% human fetal Hb in their RBC’s. IX. Cooley’s Anemia a. Also called thalassemia major or homozygous beta zero thalassemia. i. The beta zero means that they have no functional beta chains produced. b. First identified by the physician Thomas Cooley in 1925. c. The age of onset of disease is about 1 year of age, and this is after those high fetal Hb levels at birth switch to their non-functional beta globin gene. d. These patients are blood transfusion dependent from about one year of age for the rest of their lives. i. Every two weeks they come into the clinic for a transfusion. ii. With all these blood transfusions, you get a whole lot of iron from the RBC’s. 1. Our bodies do not have a way to get rid of excess iron; therefore, if you get regular blood transfusions, iron levels build. a. Iron toxicity actually causes the death of most of these Cooley’s Anemia patients in their 20s or 30s. e. Can be cured by bone marrow transplantation, but you need a histo-compatible donor, which is not readily available. i. There is a high morbidity and mortality associated with bone marrow transplants. X. Populations Affected by Beta Thalassemia [S10} a. Hemoglobinopathies and thalassemias are the most common genetic diseases worldwide. i. These diseases occur most common in equatorial regions, which are regions endemic with malaria. 1. If you are heterozygous for these disorders, you actually have a survival benefit when exposed to malaria early in life. CLASS: FUNDAMENTALS I 8-15-2011 11:00-12:00 Ryan Scribe: Daniel McCullough Proof: Joe Vaughn Hemoglobinopathies & Thalassemia Page 3 of 7 2. If you are homozygous you do not reach reproductive age, but if you are heterozygous it helps to have one of these disorders. b. There are lots of different mutations that can cause beta thalassemia. i. The whole gene can be deleted, the promoter can be deleted, upstream regulatory sequences can be deleted which will cause reduced beta globin expression. ii. You can have point mutations and splicing mutations. There are over 200 mutations that can cause beta thalassemia in humans. c. For sickle cell disease, one mutation (the same one) causes the disease every time. XI. Hemoglobin Switching During Development [S11} a. Mice do not have a fetal Hb equivalent; they make their adult globins early in development. i. Therefore, if you knock out all these mouse beta globin genes, you have a dead mouse around embryonic day 14. XII. Making Transgenic Mice by Pronuclear DNA Injection [S12] a. The trick in making mouse models, we had to give the mouse a fetal Hb. How do we do studies like this? b. We had to identify all of the regulatory sequences in the human DNA that were necessary to get those four levels of globin gene expression. i. Through making transgenic mice, we would take human bits of DNA that would have the human beta gene or the human alpha gene, and then attach regulatory sequences that we thought would increase their expression. 1. These would be tested in mice. We would take a fertilized mouse egg and take an injection pipet and inject the DNA solution into the male pronucleus. a. The pronucleus will swell up to a larger size. b. In some of the injected embryos, the DNA will integrate into the chromosome. i. If we then take the embryo and place it into a pseudo-pregnant foster mouse, when the mouse is born the DNA will be analyzed to see if it contains the injected DNA. 1. A certain fraction of the injected embryos will have the human DNA in them. These are called transgenic mice. c. Over the years, we have tested thousands of transgenic mice to see what human DNA sequences are necessary to get that high level, tissue-specific, developmentally correct expression for both the alpha and beta globin gene. XIII. GFP – Transgenic Mice [S13] a. A picture of a Green Fluorescent Protein (GFP) transgenic mouse. i. This is a protein/gene that exists in jellyfish that people have used in the lab to aid in experiments. 1. So here they just took the DNA, put it downstream of a promoter that is expressed in all cells, and made a transgenic mouse. If you shine the right wavelength of light on it, wherever the mouse doesn’t have hair you will see them fluorescing green. ii. So every cell, when you inject that fertilized egg, if your DNA goes in and you get a transgenic animal, every cell in that whole animal has your DNA in it. 1. The sequences you attach to your gene of interest determine what tissues they are expressed in. XIV. Embryonic Stem (ES) Cells [S14] a. The 2nd tool we have is embryonic stem cell technology. i. We can use this technology to manipulate and change the mouse genome any way we want. ii. We can go from an idea to a mouse on the ground in 3 months. b. The zygote that you inject DNA into to make a transgenic mouse develops to a blastocyst stage. (About 3 ½ days in the mouse). i. You can then develop two different cell types: 1. Trophoblast cells which go around the perimeter of the embryo. 2. Little ball of cells called the inner cell mass. This is where you find embryonic stem cells. a. If you take the stem cells out and plate them in a culture dish they will grow in little colonies called embryonic stem cell colonies. They will grow forever in a dish and they can be manipulated. i. You manipulate them to contain the modifications and changes that you want their DNA to have. Once you prove that the cells have been modified at the DNA level, then you can inject these cells back into a blastocyst of a wild type embryo. CLASS: FUNDAMENTALS I 8-15-2011 11:00-12:00 Ryan Scribe: Daniel McCullough Proof: Joe Vaughn Hemoglobinopathies & Thalassemia Page 4 of 7 1. Some of these cells will recombine with the inner cell mass cells and the mouse born will be a mixture, a chimera, of the wild type or black ES cells and the genetically modified white ES cells. XV. Blastocyst: Source of Embryonic Stem (ES) Cells [S15] XVI. Homologous Recombination in Embryonic Stem Cells [S16] a. How do we knock out a gene? If we want to insert a sequence to disrupt the function of a gene in region C-D: i. We insert the sequence into the gene, and make a mouse, by flanking the NEO sequence with sequences upstream and downstream of where we want homologous recombination to occur. 1. These are called homology arms or homologous sequence. 2. If homologous recombination occurs between this sequence and the chromosomal sequence, then this NEO gene will be inserted into the chromosome. XVII. Slide 16 a. If you did this for the mouse beta globin genes: i. Instead of just inserting a sequence, we took sequences way upstream and downstream of the adult gene, injected that, and got recombination, which deleted both adult genes. This caused a 16-kilobase deletion, removing the two adult genes. b. We did that in the ES cells, injected a normal host blastocyst, made a chimeric mouse and then bred it which produced mice that are all derived from your targeted ES cells. i. This would be a heterozygous beta globin knock out mouse. It only has half the normal amount of mouse beta globin genes, makes half the amount of beta globin protein, and has beta thalassemia. XVIII. Human gamma beta A Knock-in [S18] a. By using transgenic mice and ES cell technology, we figured out what sequences were necessary to give us high level fetal and adult human globin gene expression. i. Using ES cell technology, we made a construct with sequence upstream and downstream. 1. In between these homology arms we inserted human fetal gamma globin gene and a human adult beta globin gene. b. The chromosome, after successful gene targeting, would have the embryonic gene still, but also a human fetal and human adult gene. i. If we analyze these mutated embryos through development, the early mouse embryo would still make their embryonic beta H1, which switches to epsilon Y. This epsilon Y would then switch to gamma, the human fetal gene, and then the human fetal would switch to the adult beta. This would give the mouse human fetal Hb. XIX. Fetal to Adult Hb Switching in Human gamma beta A Globin Knockin Mice [S19] a. This graph analyzes the gene expression of these genes in mouse embryos over time. The X-axis is embryonic ages. i. We saw that the mouse embryos made the mouse embryonic beta H1. Then it switched off to the mouse embryonic epsilon Y. It switched off to the human fetal through most of fetal life, and then it switched off about the time of birth to the adult beta. This gives the mouse a human fetal Hb. XX. Cooley’s Anemia Mouse Model [S20] a. Now we are ready to make a Cooley’s Anemia mouse model. i. We can now keep these homozygous embryos alive during fetal life on human fetal Hb. ii. We repeated the experiment, except now we inserted a mutation in the human beta globin gene so that no functional beta globin chains would be produced. 1. We inserted it in place of the two adult mouse genes and then made heterozygous ES cells and made mice from those. XXI. Heterozygous Human gamma beta 0 KI Mouse Model [S21] a. The two left panels are what the peripheral blood looks like in a heterozygous human gamma beta 0 knock-in into the mouse locus. i. Wild-type mice have the normal looking RBC’s. ii. The beta thalassemic cells have less Hb per cell, they have a shorter half-life, and they are misshapen. 1. By electron micrograph, you can see inclusions in them which are xs?? alpha globin chains. a. These cells have too much alpha, they don’t have beta to make dimers and tetramers. b. Therefore, these xs?? alphas precipitate and cause the shortened half-life of these thalassemic RBC’s. b. This is what we saw in a heterozygous human gamma beta 0 knock in. i. Unfortunately, when we bred these to homozygosity, the animals died right before birth (18-19 days out of the 21 days of gestation). CLASS: FUNDAMENTALS I 8-15-2011 11:00-12:00 Ryan Scribe: Daniel McCullough Proof: Joe Vaughn Hemoglobinopathies & Thalassemia Page 5 of 7 1. This is longer than the 14 days that we see if you just delete the mouse genes. The presence of the gamma human fetal gene kept the animals alive for another five days, but they are still dying two days before birth. 2. We didn’t have any human alpha, so they are not making human fetal Hb. a. They are making a hybrid mouse-human Hb. Mouse alpha is pairing with human gamma. i. This hybrid Hb has an extremely high oxygen affinity. Therefore, the animals couldn’t survive beyond 18 days of gestation. XXII.Human α2α1 Globin Knock-in [S22] a. Next, we put human alpha at the mouse alpha globin locus. i. We replaced the adult mouse alpha globin genes with human alpha. ii. We put in the two human adult alpha globin genes, alpha 2 and alpha 1. 1. The final mouse alpha globin locus would have the mouse embryonic zeta gene, which would be made in the early embryonic cells of the yolk sak, and then the adult genes would come on for the remainder of the animal’s life. 2. When we bred this mouse with the gamma beta 0 knock in mouse, the animals actually make human fetal Hb (human alpha 2, human gamma 2). These mice survived. XXIII. Humanized Cooley’s Anemia Mice Survive at Birth on Human Fetal Hemoglobin [S23] a. Inside the RBC’s of a humanized Cooley’s Anemia mouse, they have 100% human fetal Hb. There is no mouse globin anymore. i. HPLC traces show that in the Cooley’s Anemia knock in mouse, they have human alpha and human gamma. 1. If you have a normal beta globin gene in there, they will make alpha, a little bit of gamma, and beta right at birth. Then the gamma will continue to switch until you have nothing but beta later on. (NOTE TO READER: THIS IS NOT WHAT HAPPENS IF THERE IS ONLY HUMAN ALPHA AND HUMAN GAMMA IN THE MOUSE. THE NORMAL BETA GLOBIN GENE IS REFERRING TO A MOUSE THAT STILL HAS THEIR OWN ADULT MOUSE ALPHA GLOBIN GENES!!) a. This also occurs in the knock in mice. They continue the switch, and as they switch they have Cooley’s Anemia; therefore, unless we give them blood transfusions they die. ii. So here we have a model that looks just like the human condition. 1. They have high levels of human fetal Hb at birth, and then as the switch is completed they have no beta chains. XXIV. Hemoglobin Switching in Humanized γHPFHβ0 and γHPFHδβ0 Globin Knockin Mice [S24] a. These Cooley’s Anemia model mice actually died a little earlier than we wanted them to, so we manipulated them one more time to make fetal Hb levels last longer after birth. i. In humans, there are mutations called hereditary persistence of fetal Hb mutations (HPFH), and these are mutations in the promoters of the human fetal gene. 1. Patients that have these have higher levels of fetal Hb in the adult. 2. We tested to see if these same mutations would work in the mouse RBC. They did work. a. We used an HPFH mutation 117 nucleotides upstream in the human gamma globin promoter. We put in the mutation that occurs in the human patients. i. This resulted in higher human fetal Hb levels in the mice after birth. 1. At birth, wild type gamma genes have about 40% gamma chains. 2. When we put in an HPFH mutation, that 40% went up to about 70%, which is what we see in newborns. 3. In another mouse, we inserted the minor adult delta globin gene along with the HPFH mutation. a. This causes delta to come on and go up to about 5%, gamma goes down to about 5%, and beta makes up the rest. This is very similar to what occurs in humans. XXV. Survival Curves of Humanized Cooley’s Anemia Mice [S25] a. If we made the mice homozygous, where they can’t make any adult beta globin but only the fetal, they survived about two weeks. i. The survival curves show 100% survival at birth. However, about two weeks after birth we lose a majority of the mice. CLASS: FUNDAMENTALS I 8-15-2011 11:00-12:00 Ryan 1. XXVI. XXVII. XXVIII. XXIX. XXX. XXXI. XXXII. XXXIII. Scribe: Daniel McCullough Proof: Joe Vaughn Hemoglobinopathies & Thalassemia Page 6 of 7 A two week old mouse is about a 1 to 1 ½ year old human. In Cooley’s Anemia patients, unless they get blood transfusions, they would expire at about 1 ½ years of age. Now we have the timing just like it is in human patients. Sickle Hemoglobin [S26] a. Sickle Cell Disease is caused by a single AA change in the 6th position of the beta chain. A glutamic acid is changed to a valine. i. In the DNA, there is a nucleotide change which causes the codon to code for a valine instead of a glutamic acid. ii. Valine is hydrophobic and is right on the surface of the globin, which means it is exposed to water. It doesn’t like to be there, so it looks for a place to hide. Sickle Cell Anemia [S27] a. 1st identified by the physician James Herrick in 1910. b. The presence of the AA change causes a hemolytic anemia. i. These RBC’s have a very short half-life and they lyse. c. Sickle Cell Anemia causes the RBC’s to be misshapen which can occlude the vasculature. i. This causes very severe painful crises. The tissue downstream of the occluded vessel receives no oxygen, which is sensed as pain. d. Sickle Cell Anemia patients have major problems such as stroke and splenic sequestration i. Splenic sequestration is when sickled blood goes into the spleen and blocks the outflow of the blood from the spleen. The blood will keep going in without the blood exiting. This causes the spleen to enlarge. 1. This is life threatening. e. Can be cured by bone marrow transplantation. Sickle-Cell Anemia is a Molecular Disease [S28] a. Sickle Cell Anemia patients have abnormally shaped RBC’s. The erythrocytes are crescent shaped only in the deoxy form of the RBC. i. In sickle Hb, or Hb S, the Hb tetramers don’t aggregate and form sickle fibers if in the oxygenated form. ii. Only when HB goes to the terse, or deoxy, form do all the disease consequences happen. b. The result of the glutamic acid being replaced by valine is that the sickle molecules aggregate into long, chain-like polymeric structures. Sickle-Cell Anemia is a Molecular Disease [S29] a. The figure is a picture from your book showing oxyhemoglobin A and S and deoxyhemoglobin A and S. i. When normal Hb goes to the terse, or T, conformation, which is the deoxy form, there is a hydrophobic pocket that opens up on the beta chain. ii. In the oxyhemoglobin S, there is no hydrophobic pocket because it is in the R, or oxyhemoglobin, form. 1. It has a protrusion, which represents the valine sticking out into the polar environment of water. a. In the deoxyhemoglobin S, the valine sticks out and the hydrophobic pocket is present. b. If the valine fits into a hydrophobic pocket on a neighboring molecule, it finds a place to hide. This forms aggregates of sickle Hb. Sickle-Cell Anemia is a Molecular Disease [S30] a. In the deoxy form, where you have a hydrophobic pocket and a valine extension, you can get a polymer formed. i. In your post-capillary venules, once the partial pressure of oxygen gets low and you have a larger proportion of Hb in the deoxy form, then fibers start to form. 1. If the fibers can make it back to the lungs and get exposed to oxygen, they will fall apart and become normal. However, if they get too large before making it back to the lungs, they will occlude a vessel. 2. As a sickled RBC goes from the lungs to the tissues, it is undergoing shifts from fiber formation to no fiber formation. 3. These fibers formed can destroy the membrane and cause the early destruction of the RBC’s. Sickle Hemoglobin Polymerizes [S31] a. The fiber that is formed is a complicated structure. It has 14 strands. i. You get 7 pairs of Hb molecules that connected at the valine protrusion and the hydrophobic pocket. 1. This is a higher ordered structure called a sickle fiber. You can see this form inside the RBC in the deoxy state. Vascular Occlusion of DeoxyHbS [S32] a. (SEE SLIDES 27 AND 28. IT IS THE SAME INFORMATION THAT HE TALKS ABOUT HERE) Mouse Model of Sickle Cell Disease [S33] CLASS: FUNDAMENTALS I Scribe: Daniel McCullough 8-15-2011 11:00-12:00 Proof: Joe Vaughn Ryan Hemoglobinopathies & Thalassemia Page 7 of 7 a. To generate a mouse model of sickle cell disease we had to produce high levels of sickle Hb (human alpha and human beta S), we had to get rid of all the endogenous alpha and beta globins of the mouse. i. This is because the mouse alpha and beta have anti-sickling properties. They will not participate in the fiber formation. Therefore we needed RBC’s that had 100% human sickle Hb. ii. This mouse is called a knock out transgenic sickle mouse. XXXIV. First Generation Animal Model of SCD [S34] a. Our first generation model was just a transgenic model. It had very little in vivo pathology because of the mouse alpha and beta globins that were there. i. But, if you took the RBC’s out and deoxygenated them, you could see the fibers and the misshapen cells form. XXXV. Mouse βmajβmin Globin Knock-Out [S35] a. Then we knocked out the mouse beta and alpha. XXXVI. Cloned βThalassemic Mice [S36] XXXVII. Mouse Model of Sickle-Cell Disease [S37] a. We took the sickle mice that had high levels of alpha. We had to put the fetal gene in to keep them alive during fetal life because of beta thalassemia. We also put the beta sickle gene (βS) in. i. Then we bred these mice to alpha knock out mice and to beta knock out mice. After many generations, we got a knock-out transgenic sickle mouse that has only human sickle Hb. 1. This mouse switches from human fetal to human sickle after birth. XXXVIII. Knockout-Transgenic Sickle Mouse Blood [S38] a. The peripheral blood smear of these mice show sickled cells. XXXIX. Sickle Mouse Splenomegaly [S39] a. The spleen in the sickle mouse is quite a bit larger than the normal spleen. i. This occurs in response to the severe anemia. The spleen becomes an erythroid organ, and gets a lot of erythroblasts to make more and more RBC’s to compensate for the anemia. XL. Sickle Mouse Survival Curves [S40] a. The survival curve of the sickle mice shows that the average sickle mouse lives about a year. i. A wild type mouse can live about 2 ½ years. b. If we cross the mice that have the knockouts in the transgenic locus back to an inbred strain generation after generation, then the animals get very sick. i. They only live a month or two. XLI. Cell Therapy [S41] a. Now that we have the model for Cooley’s Anemia and Sickle Cell Disease, what do we do with them. i. We use these models to work out different stem, progenitor, and cell therapies to cure the mice. 1. To do this, we establish a cell line from the model mice, correct the mutation, and then replace the diseased cells in the mice with the corrected cells. XLII. Cell Therapy for Hemoglobinopathies [S42] a. This is basically how it would work: i. We would have a beta thalassemia or sickle mouse and take some skin cells. 1. We use the skin cells to grow out some fibroblasts in a dish. 2. Then we can express some transcription factors, which will reprogram these cells from a skin cell into a pluripotent stem cell. a. This is called an induced pluripotent stem cell, or an iPS cell. 3. Once we have the iPS cells in a dish, then we repair the lesion. a. We correct the mutation by homologous recombination. The iPS cells grow just like embryonic stem cells. 4. Once we have the repaired cells, then we differentiate them in a dish in vivo to hematopoietic stem cells. 5. Then, once we have the corrected hematopoietic stem cells, we can transplant them back into the diseased animal. a. This corrects their disease. (40:39)