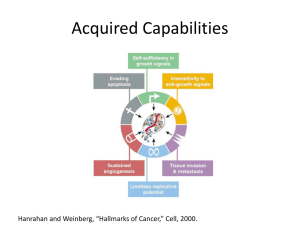
Inflammasomes in non-immune cells
advertisement

Inflammasomes in non-immune cells: functions and role in disease Author: Casper Berger, University of Utrecht, master student Infection and Immunity. Daily supervisor: MSc. Lieneke Bouwman,, University Utrecht, Fac. of Veterinary Medicine, Dept. of Infectious Diseases and Immunology. Examiner: Prof. Jos van Putten, University of Utrecht, Fac. of Veterinary Medicine, Dept. of Infectious Diseases and Immunology. Second reviewer: Dr. Hélène Verheije, University of Utrecht, Fac. of Veterinary Medicine, Dept. of Pathobiology Abstract Inflammasomes are intracellular protein complexes that consist of a molecular sensor, an ASC adaptor protein and the cysteine protease caspase-1. Inflammasomes are involved in cleaving pro-IL1β and pro-IL-18 into their active forms. Numerous studies show that beside immune cells, nonimmune cells can express functional NLRP1, NLRP3, NLRC4, NLRP6, AIM-2, RIG-I and IFI16 inflammasomes. The function of inflammasomes in non-immune cells is still largely unknown, but the inflammasome may be involved in several diseases including infections by pathogens, allergic reactions, age-related macular degeneration, ischemia and reperfusion injury, diabetes and cancer. Whether inflammasome activation is beneficial or detrimental to the development of these diseases is often still unclear. This review gives an overview of the characteristics of inflammasomes in nonimmune cells and discusses their function in comparison to inflammasomes in immune cells. Table of contents Abstract ............................................................................................................................................ 1 1. Inflammasome structure, function and activation ............................................................. 2 Structure of inflammasomes .......................................................................................................... 2 Functions of inflammasomes ......................................................................................................... 4 Mechanisms of inflammasome activation ...................................................................................... 5 Regulation of inflammasome activity ............................................................................................ 5 2. Pathogen and microbiota induced inflammasome formation in non-immune cells........ 6 Pathogens ....................................................................................................................................... 6 Microbiota ...................................................................................................................................... 7 3. (Hyper) response to foreign biological molecules ............................................................... 8 4. Response to endogenous proteins, metabolites and danger signals .................................. 9 Hyperhomocysteinemia and psoriasis ............................................................................................ 9 Glucose and the NLRP3 inflammasome ........................................................................................ 9 The NLRP3 inflammasome and age-related macular degeneration ............................................. 10 Ischemia and reperfusion injury ................................................................................................... 11 Atherosclerosis ............................................................................................................................. 11 5. Response to non-biological substances and stimuli .......................................................... 12 6. Inflammasomes in wound healing, tumor development and pain sensitivity ................ 13 Wound healing and tumors .......................................................................................................... 13 1 Pain sensitivity ............................................................................................................................. 14 Overview of inflammasome types in non-immune cells ............................................................. 16 7. Discussion............................................................................................................................. 18 References ....................................................................................................................................... 19 1. Inflammasome structure, function and activation Inflammasomes are intracellular protein complexes involved in the activation of inflammatory cytokines in response to pathogens and danger signals, resulting in inflammation and/or cell death. This makes inflammasomes important players in the innate immune response. The primary focus of studies on inflammasomes so far has been their role in dedicated immune cells of hematopoietic origin. However, the molecular components of inflammasomes are also present in non-immune cells. This review focuses on the role of inflammasomes in non-immune cells. In the first section an introduction on the inflammasome structure and the different inflammasome types is provided. The second part gives an overview of the roles of inflammasomes in diverse processes in non-immune cells including the response against pathogens, ischemia and reperfusion injury, metabolic disorders, auto-immune diseases, allergic reactions and cancer. Finally I will discuss the relative role of inflammasomes in non-immune cells compared to immune cells. An overview of studies on inflammasomes in non-immune cells is provided in Table 1. Structure of inflammasomes The primary function of inflammasomes in immune cells is to trigger inflammation and cell death in response to pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs). This is primarily mediated through the processing of pro-IL-1β and pro-IL-18 into their inflammatory active form. Canonical inflammasomes consist of three components to accomplish this: a sensor molecule, the adaptor protein apoptosis-associated speck-like protein (ASC) and the cysteine protease caspase-1 (Figure 1). The sensor molecule binds to adaptor protein ASC with its pyrin domain when activated by PAMPs and DAMPs. ASC in turn binds monomeric pro-caspase-1 via its caspase recruitment domain (CARD). This results in auto-cleavage of pro-caspase-1 into the active caspase-1 which subsequently forms heterotetramers of the P10 and P20 caspase- subunits. The caspase-1 in the protein complex then cleaves the interleukins pro-IL-1β and pro-IL-18, resulting in release in their activated form[1]. Molecular sensor ASC P20 pro-IL1-β 1β 2 P10 IL1-1β Figure 1. Schematic overview of inflammasome formation. A molecular sensor is directly or indirectly activated by PAMPs and DAMPs, facilitating subsequently binding to the adaptor protein ASC. ASC then binds procaspase-1 which results in auto-cleavage into the P10 and P20 subunit. Heterotetramers of the P10 and P20 subunit are formed which cleave pro-IL1β and pro-IL-18. Different inflammasomes can be formed depending on the type of molecular sensor that is involved. Many of the molecular sensors are NOD like-receptors (NLRs). Most NLRs involved in inflammasome formation including NLRP3, NLRP6 and NLRP7 contain a pyrin domain with which they can bind to ASC. However, some NLRs lack a pyrin domain. NLRC4 contains a CARD instead of pyrin domain and can directly interact with caspase-1. Although ASC is not required for pro-IL-1β processing, it does enhance it[1]. Human NLRP1 contains both a pyrin and a CARD domain whereas murine NLRP1-a and –b (mice have three NLRP1 isoforms) only contains a CARD domain. NLRP1c has a truncated CARD domain[2][3]. Although human NLRP1 has a CARD domain and can therefore directly interact with pro-caspase-1, ASC has been shown to enhance NLRP1 inflammasome activity in humans[2]. All NLRs also contain a NAIP, CIITA, HET-E and TP1 (NACHT) domain which is involved in ATP-dependent oligomerization with other NACHT domain containing proteins, facilitating the complex formation. They also have C-terminal leucine-rich repeats which have regulatory functions[1].The non-NLR proteins retinoic acid-inducible gene I (RIG-I), interferon gamma-inducible protein 16 (IFI16) and absent in melanoma-2 (AIM-2) can also act as molecular sensors and form inflammasomes[1][4]. IFI16 and AIM-2 contain one or more HIN domains which bind double stranded DNA[1]. RIG-I can also bind to double stranded DNA[4]. Off all the known inflammasome types, the NLRP3 inflammasome is the best characterized. An overview of inflammasome formation is provided in Figure 2. Figure 2. Schematic overview of NLRP1, NLRP3, NLRC4 (IPAF) and AIM-2 inflammasomes. NLRP1 and NLRC4 contain a CARD domain and can directly interact with caspase-1 (although ASC enhances NLRP1 and NLRC4 inflammasome activity[1][2]). NLRP3and AIM2 bind ASC with their pyrin domain which in turn binds to caspase-1 via its CARD domain. Binding of the molecular sensor directly or via ASC to caspase-1 results in caspase-1 auto-cleavage yielding P20 and P10 subunits (cleavage sites indicated with arrows). These can form heterotetramers which can cleave pro-IL-1β. Image originally published by K Schroder and J Tschopp 2010[2]. Variations in inflammasome components are not limited to the sensor proteins, but also exist in the ASC protein and the protease. Three different isoforms of ASC are known to exist (ASC –a, - b and – c). ASC-b lacks the linker domain between the pyrin and CARD domain. ASC-b has been shown in vitro to be able to form functional NLRP3 inflammasomes while ASC-c lacks parts of the pyrin domain but still has a functional CARD domain. ASC-c has been shown in vitro to inhibit inflammasome formation, possibly by competing for binding sites on pro-caspase-1 with conventional full-length ASC-a[5]. ASC has also been reported to have tumor repressive effects independent from inflammasomes by activating P53[6]. 3 Activation of pro-caspase-1 auto cleavage can also occur through murine caspase-11 (caspase-4 and/or 5 in humans). Caspase-11 may be activated by TLR4 and the downstream TRIF pathway. Activation of this pathway results in interferon -α and -β release, which results in pro-caspase-11 auto cleavage and subsequent pro-caspase-1 auto-cleavage of the NLRP3 inflammasome. Pro-caspase-11 is also auto-cleaved when expressed at sufficiently high cellular levels[7]. The mechanism by which caspase-11 affects NLRP3 inflammasome activation is not fully understood[8]. Functions of inflammasomes The primary function of inflammasomes appears the processing of pro-IL-1β and pro-IL-18 and their subsequent release by non-classical protein secretion (because IL-1β and pro-IL-18 lack a signaling peptide). IL-1β and IL-18 promote inflammation by recruiting immune cells, inducing fever, increasing pain sensitivity and vasodilation[9]. It should be noted that IL-1β has been reported to be secreted in extracellular membrane vesicles[10] beside being released as a soluble proteins. Proteins present in extracellular vesicles cannot be detected with assays such as ELISA unless the vesicles are lysed and may therefore be missed by assays. Inflammasome-independent cleaving of pro-IL-1β, pro-IL-1β can also occur by a protein complex that consists of mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALTI1), ASC and caspase-8 can also process pro-IL-1β. This complex can be activated by specific pattern recognition receptors such as dectin 1, the ripoptosome (a protein complex involved in cell death in response to genotoxic stress) and activation of the tumor necrosis factor receptor (TNFR) CD95[1]. Beside IL-1β, the interleukin IL-1α, which also binds the IL-1 receptor, can trigger inflammation. Whether IL-1α processing and release are also controlled by inflammasomes is less clear. The fulllength IL-1α protein can be cleaved by a calpain-like protease, but unlike IL-1β both the cleaved and the non-cleaved form are biologically active. IL-1α cleavage and release may be partially dependent on the inflammasome and caspase-1 (but not the protolithic activity of caspase-1). IL-1α may not be actively released at all and rather act as an inflammatory signal when it leaks from cell dying due to non-apoptotic cell death[11]. However, Lee et al. 2009 report that a caspase-1 inhibitor reduced IL-1α release from the epidermis[12]. Inflammasomes are also involved in pyroptosis. Pyroptosis is a programmed cell death pathway distinct from apoptosis and dependent on caspase-1. Pyroptotic cells swell resulting in rupture of the plasma membrane and the release of inflammation inducing molecules. Caspase-1 and caspase-11 are involved in pyroptosis and inflammasome formation can result in pyroptosis. Multiple inflammasome types such as NLRP3, NLRC4 and AIM-2 can initiate pyroptosis[9][13]. In addition to interleukin processing and pyroptosis, inflammasomes may have additional functions which are relatively unexplored. One study identifies 41 substrates of caspase-1 by using a diagonal 2D gel with in-gel caspase-1 digestion followed by identification of digested proteins with mass spectrometry. The identified substrates are involved in diverse processes including the cytoskeleton and energy metabolism. Five of the identified substrates are cytosolic enzymes in the glycolysis pathway. Some of these enzymes are cleaved in mouse macrophages in response to Salmonella infection in vitro and this is dependent on caspase-1. Additionally, lactate levels are higher in Salmonella infected macrophages isolated from caspase-1 knockout mice compared to macrophages isolated from wildtype mice. This may suggest that caspase-1 activation reduces glycolysis[14]. In a different study, caspase-1 activation by pore forming toxins results in a ASC, NLRP3 and NLRC4 -dependent sterol regulatory element binding protein -1 and -2(SREBP) activation which in turn results in increased cell survival of HeLa cells[15]. SREBPs are transcription factors that play a central role in membrane biogenesis. How caspase-1 activates SREBPs has not been established. Interestingly, SREBP-1a has also been shown to directly stimulate NLRP1a transcription but not of 4 NLRP1c[16]. These studies show that caspase-1 has more functions beside the processing of pro-IL1β and pro-IL-18 and pyroptosis. They also indicate that inflammasome activation may have more diverse functions than interleukin processing and pyroptosis. Mechanisms of inflammasome activation The sensor molecules of inflammasomes can directly detect distinct signals such as the bacterial cell wall component muramyl dipeptide by NLRP1 and double stranded DNA by AIM-2. The NLRP3 inflammasome on the other hand can be activated by a wide range of stimuli including pathogens, metabolites, many inorganic crystalline compounds and UVB. Several reports suggest that an efflux of K+ is a central mechanism in NLRP3 inflammasome formation and may help explain the broad specificity. An efflux of K+ has been shown to result in reactive oxygen species (ROS) formation and subsequent NLRP3 inflammasome dependent caspase-1 activation[17]. Permeabilization of the plasma membrane by pore forming bacterial toxins also results in a K+ efflux and subsequent inflammasome activation[15]. Additionally, the phagocytosis of crystalline compounds such as urate crystals can result in ROS formation by damaging lysosomes resulting in NLRP3-inflammasome formation[2]. A more recent study suggests that although a K+ efflux is essential for NLRP3 inflammasome activation, ROS formation is not critical to the process[18]. The leaking of lysosomal enzymes into the cytosol such as cathepsin B has also been implicated in NLRP3 inflammasome activation[19]. Beside inflammasome activation, upregulation of pro-IL-1β is also required. Pro-IL-1β is not constitutively expressed in most cell types but instead is upregulated by NF-κB in response to NOD and Toll-like receptor activation. Regulation of inflammasome activity The activity of the inflammasome is controlled at multiple levels. Beside the three core-components described above the formation of some inflammasomes may require the involvement of co-receptors. NLRC4 may require NAIP family members for inflammasome formation. NAIP family members can detect proteins of pathogens such as flagellin by NAIP5 and NAIP6 and a type III secretion system component is detected by NAIP2 in mice. Only a single NAIP orthologue is known in humans though. The formation of the NLRP1inflammasome is likely dependent on NOD2[1][2]. For NLRP3, activity is controlled at both the transcriptional and protein level. NLRP3 expression is low under normal conditions and needs to be upregulated to increase inflammasome formation. Additionally, deubiquitination of NLRP3 by the deubiquitinase BRRC3 enhances NLRP3 activity[1]. Autophagy may play a role in limiting inflammasome activation by preventing accumulation of factors that stimulate inflammasome formation, such as ROS and mitochondrial DNA in the cytoplasm[20]. Negative regulation of NLRP3 inflammasomes has been shown to occur by direct contact of CD4+ T cells and is possibly dependent on a CD40 ligand[1][2]. Many more factors that regulate inflammasome formation have been described but it is outside the scope of this review to describe them all. An excellent in-debt review on regulation of inflammasomes has been written by Latz et al. 2013[1]. Inflammasomes and their function appear to be more complex as originally described. More sensor molecules continue to be identified and some can also directly bind to pro-caspase-1 rather than to ASC. Inflammasomes components are regulated at multiple levels by a variety of factors. Alternative inflammasome activation pathways have been identified and inflammasome independent processing of IL-1β can also occur. Regarding their functions, beside their well-known role in interleukin processing and pyroptosis, inflammasomes have been shown to affect membrane biogenesis and a 5 large number of substrates have been identified for caspase-1 including glycolysis enzymes. These additional levels of complexity may help to explain some of the observed discrepancies between different studies and underline the importance of the more biologically relevant in vivo models compared to in vitro models to verify findings on inflammasomes. They also make it more difficult to define what an inflammasome is and what is not. ASC is not strictly required and neither is interleukin processing its only function. Pro-caspase-1 activation, a sensor molecule that recognizes PAMPs and/or DAMPs and the formation of a protein complex appear indispensable for canonical inflammasomes. However, caspase-8 dependent IL-1β processing can also occur. This complex shares many properties with canonical inflammasomes and can therefore arguably be termed a type of inflammasome as well. What both complexes do have in common is that they are activated by stimulation of PAMP- and DAMP-sensing molecules, form a large protein complex that results in the activation of caspase-1 or caspase-8 which in turn can cleave pro-interleukins to activate them. 2. Pathogen and microbiota induced inflammasome formation in non-immune cells Many pathogen components are directly or indirectly recognized by the molecular sensors, resulting in inflammasome activation and interleukin processing. The majority of studies focus on this role in immune cells, but some studies also show inflammasome activation in response to pathogens in nonimmune cells. Beside recognizing pathogens, effects of inflammasomes on the composition of the microbiota and resulting pathologies have also been reported[21][22]. Pathogens One pathogen that has been shown to trigger inflammasome activation in non-immune cells is the influenza A virus. Infection by influenza A virus results in a RIG-I and NLRP3 inflammasome dependent IL-1β release by primary human lung epithelial cells in vitro[4]. RIG-I activation also resulted in RIG-I, TLR3 and NLRP3 upregulation. This upregulation depends on interferon-β and the type I interferon receptor 1 α chain (IFNAR1), which increases RIG-1, TLR3 and NLRP3 expression[4]. These results show the direct formation of NLRP3 and RIG-I inflammasomes in response to a virus and an indirect role for RIG-I in NLRP3 inflammasome activation in non-immune cells. The authors did not compare the IL-1β release to the amount of IL-1β released by macrophages or other immune cells from the same donors, so the relative contribution to IL-1β release remains currently unknown. IFI16 inflammasome activation has also been implicated in response to Kaposi's sarcomaassociated herpesvirus (KSHV)[10]. Increased caspase-1 activation and processing of IL-1β were observed in latently infected endothelial telomerase-immortalized human umbilical cells (TIVE) and B-cells but not in uninfected cells. Co-immunoprecipation experiments showed that IFI16 forms protein complexes with ASC and caspase-1. Fluorescence microscopy revealed that in non-infected cells IFI16 is mostly localized in the nucleus but localizes mostly perinuclear with ASC during KSHV infection. FISH staining of the KSHV genome shows that it colocalizes with IFI16, suggesting that it is directly detected by IFI16[10]. KSHV normally infects B-cells in vivo so it remains to be seen whether KSHV also results in inflammasome activation in non-immune cells in vivo. Yersinia enterocolitica infection also triggers inflammasome activation[23]. Y. enterocolitica infection of Caco-2 cells results in NLRP3 inflammasome-dependent IL-18 release triggered by a K+ flux. No IL1β release was observed however. Binding of the bacterial adhesin invasin (Inv) to α5β1 integrin resulted in increased IL-18 release during infection due to increased IL-18 expression and is therefore an alternative priming signal[23]. Inflammasome activation in non-immune cells in response to pathogens is not necessarily beneficial for the host. An in vitro study shows that Chlamydia trachomatis triggers NLRP3 6 inflammasome formation in HeLa cells[17]. The type III secretion system is required for this activation. The type III secretion system triggers a K+ efflux which results in ROS formation and subsequent NLRP3 inflammasome formation and caspase-1 activation. Interestingly, C. trachomatis benefits from the observed inflammasome activation since inhibition of caspase-1 results in decreased infection[17]. The bacterium Pseudomonas aeruginosa does not appear to stimulate inflammasome activation in non-immune cells whereas it does in immune cells. One study observed hardly or no caspase-1 activation and IL-1β release of 4 different lung epithelial cell lines in response to P. aeruginosa whereas blood mononuclear cells did show caspase-1 activation and IL-1β release[24]. Another study compared IL-1β release from macrophages and the total lung tissue in response to P. aeruginosa[25]. They observed that the majority of IL-1β is released by macrophages and is caused by activation of the NLRC4 inflammasome by the bacterial flagellin. Interestingly, inflammasome activation was associated with more severe disease outcome in mice. Mice challenged with wildtype or a mutant P. aeruginosa with nonfunctional flagella showed decreased survival and more tissue damage compared to infection with a mutant P. aeruginosa that lack flagellin[25]. Microbiota Two studies using knockout mice show that the NLRP3 and NLRP6 inflammasomes activity in nonimmune cells have an effect on the microbiota[21][22]. The changes in microbiota resulted in inflammatory pathology. In one study, NLRP6, ASC and caspase-1 knockout and wt mice were given dextran sodium sulfate (DSS) which is known to result in inflammation of the colon (colitis). The knockout mice developed more severe colitis compared to wt mice, indicating a protective role of NLRP6 inflammasomes. This phenotype was transferable to co-housed wt mice and treatment with antibiotics reversed the more severe colitis of the knockout phenotype, suggesting that the phenotype is caused by changes in microbiota caused by deficient inflammasomes. This effect was caused by IL18 rather than IL-1β as shown by knockout experiments. The changes in microbiota were primarily caused by the NLRP6 inflammasome in non-immune cells as shown by knockout experiments with adoptive transfer of IL-18 and NLRP6 knockout bone marrow[22]. Another study with very similar experiments indicates that the NLRP3 inflammasome likely plays a role in non-alcoholic steatopepatitis (NASH). ASC, caspase-1 and NLRP3 knockout mice fed a methionine-choline-deficient diet (which is known to induce NASH) have increased NASH symptoms compared to wt mice. The more severe NASH phenotype was shown to be caused by changes in the microbiota with co-housing experiments and antibiotic treatment. Adoptive bone-marrow experiments confirmed that inflammasome activation in non-immune cells is responsible for the altered NASH phenotype. The altered microbiota results in an increase of TLR4 and TLR9 agonists reaching the liver through the portal vein, which triggers inflammation in the liver[21]. However, it was not investigated how the altered microbiota results in more TLR4 and TLR9 ligands reaching the liver through the portal vein. Activation of NLRP6 inflammasome in goblet cells and epithelial cells has also been shown to reduce mucus secretion[26]. This effect was caused by NLRP6 inflammasome induced reduction of autophagy which in turn results in deficient mucus secretion and accumulation of mucus-containing vesicles on the apical side of goblet cells[26]. Collectively, these studies show that inflammasomes in non-immune cells can have a role in response to pathogens and commensal microbiota. The relative contribution of inflammasomes in non-immune cells to the response against pathogens is difficult to determine. The variable activation of inflammasomes observed in non-immune cells in response to different pathogens [25][24][4][17] may indicate that inflammasome activation in non-immune cells is dependent on a number of factors such 7 as the host immune system and the type of pathogen. Although IL-1β release of immune cells seems higher than non-immune cells, the IL-β release by non-immune cells may still play a role in initiating inflammation by recruiting and activating innate immune cells which can then further amplify the immune response. The relatively much greater number of non-immune cells compared to the number of immune cells in peripheral tissues should also be considered. Autocrine activation of surrounding epithelial tissue may amplify the immune response. Epithelial cells are also the first cells to be exposed to a pathogen which would make them good candidates to initiate an immune response. Apart from interleukin processing and release, alternative functions of inflammasome activation in non-immune cells should be considered such as pyroptosis and alterations in metabolism. These may have a more prevalent role than interleukin processing in non-immune cells. 3. (Hyper) response to foreign biological molecules Inflammasome activation in non-immune cells can also occur in response to non-pathogen derived foreign biological molecules. Several studies implicate a role for NLRP3 and AIM-2 inflammasomes in keratinocytes in the development of allergic skin responses. ASC, NLRP3 and IL-1R knockout mice sensitized with dinitro-1-fluorobenzene (DNFB) or trinitrochlorobenzene (TNCB) on the skin and later -re-exposed to the same compound showed reduced inflammation compared to wildtype in the early phase of inflammation, and also in the late phase in IL-1R knockout mice[28]. Furthermore, it was demonstrated that in vitro mouse keratinocytes release IL-1β in response to various stimuli including sodium dedecyl sulfate (SDS), UVB (UVA to a lesser extend) and DNFB. The latter was used to trigger inflammation in the mouse model and therefore suggests that inflammasome activation in keratinocytes is likely to play a role in vivo as well[28]. In another study, inflammasome activation with DNFB has been shown to result in IL-1β release and a decrease in expression of thymic stromal lymphopoietin (TSLP) in mouse and human keratinocytes. TSLP skews an immune response toward a Th2 response which is associated with more severe symptoms to allergens in atopic dermatitis and asthma[29]. The type(s) of inflammasome(s) involved was not determined in this study. It should be noted that contrary to immune cells, pro-IL1β is constitutively expressed in keratinocytes[27] A role for NLRP3 inflammasomes has also been suggested in atopic dermatitis. The potent dust mite allergen Der p 1 has been shown in vitro to activate NLRP3 inflammasomes in keratinocytes. IL1β and IL-18 processing and release were increased by activation of the NLRP3 inflammasome by Der p 1 whereas IL-1β and IL-18 expression levels remained unaffected. Inflammasome activation by Der p 1 depends on its protease activity since it was unable to trigger inflammasome activation after heat inactivation of the enzymatic activity. The inflammasome activation also required a K+ efflux as high levels of extracellular K+ blocked inflammasome activation[30]. The mechanism by which Der P 1 activates NLRP3 inflammasomes was not determined. A possible explanation could be that Der p 1 (partially) digests proteins on the surface of keratinocytes. A role of inflammasomes in strong immune responses against foreign biological molecules is not limited to the formation of NLRP3 inflammasomes in keratinocytes. The bee-venom component melittin triggers AIM-2 but not NLRP3 inflammasome dependent IL-1β processing in human keratinocytes. The activation of AIM-2 inflammasome was caused by a disruption of mitochondria by melittin which results in leakage of mitochondrial DNA into the cytosol which is recognized by AIM2[31]. It is puzzling however why melittin did not disrupt the plasma membrane, which would result in a K+ efflux and subsequent NLRP3 inflammasome activation. Additionally, Ca2+ release from mitochondria has also been implicated in NLRP3 inflammasome activation[1]. Overall, these studies show that when keratinocytes are directly exposed to strong allergens they can contribute to an inflammasome-dependent immune response. Both NLRP3 and AIM-2 inflammasomes have been shown to be functional in keratinocytes. However, the role of dedicated 8 immune cells cannot be excluded based on these studies and I expect them to play a role as well. Further study is required to determine their relative contribution to inflammasome-dependent immune responses in different allergic reactions. 4. Response to endogenous proteins, metabolites and danger signals Beside foreign activating signals, self-components such as proteins and metabolites can activate inflammasomes in non-immune cells and this has been implicated to play a role in several disorders including hyperhomocysteinemia, psoriasis, diabetes, age-related macular degeneration, ischemia/ reperfusion injury and atherosclerosis. Hyperhomocysteinemia and psoriasis Insufficient clearing of the metabolite homocysteine (a cysteine homologue), termed hyperhomocysteinemia, can result in kidney inflammation and damage. It has been shown that NLRP3 inflammasomes in podocytes become activated by homocysteine in a mouse model of hyperhomocysteinemia. This results in the release of IL-1β and increased tissue damage [32]. The inflammasome activation was shown to be dependent on NADPH for the generation of ROS[33]. However the dependence on NADPH for NLRP3 inflammasome activation has been challenged[1]. A role for inflammasome activation in immune cells in response to hyperhomocysteine (in addition to non-immune cells) cannot be excluded based on these results. AIM-2 inflammasomes have been shown to play a role in the inflammatory skin disease psoriasis. Increased IL-1β and activated caspase-1 were observed in skin lesions of psoriasis patients and AIM-2 protein expression levels were 50-fold increased as well. Microscopy imaging of skin tissue revealed that the majority of keratinocytes in psoriatic lesions contain cytosolic DNA whereas this was not observed for keratinocytes in healthy control skin[34]. Glucose and the NLRP3 inflammasome The NLRP3 inflammasome is known to be activated in response to high levels of various metabolites including glucose. This links the NLRP3 inflammasome to diabetes type II and associated pathology. NLRP3 inflammasome activity has been shown in both immune and non-immune cells in response to glucose. Stienstra et al. 2010 show that NLRP3 inflammasome dependent caspase-1 activity and subsequent IL-1β and IL-18 release is increased in adipose tissue of obese mice compared to the control and that this increase is likely derived from adipocytes rather than macrophages[35]. Caspase1 knockout mice have a lower body weight and smaller adipocytes compared to wildtype mice. Additionally, adipose tissue from caspase-1 and NLRP3 knockout mice is more sensitive to insulin and less sensitive to glucose compared to healthy control mice, suggesting a detrimental effect of inflammasomes on diabetes[35]. A different study reports that knockout of the inflammasome components NLRP3 and caspase-1 results in increased insulin sensitivity and glucose tolerance of adipose tissue in obese mice compared to obese wildtype mice. However, contrary to Stienstra et al. 2010[35], this study suggest a primary role of NLRP3 inflammasome activation and IL1-1β processing in macrophages rather than adipocytes in response to glucose[36]. It is interesting to note that although both studies report a large decrease of IL-1β release in NLRP3[36] and caspase-1[35] knockout mice IL-1β release is not completely ablated either. This suggests a role for canonicalinflammasome independent pro-IL-1β processing and possibly the involvement of inflammasomes with other molecular sensors[37]. Mouse islets cells have also been shown to release IL-1β in response to high levels of glucose in vitro. This release is largely dependent on NLRP3 and thiorexodin-interacting protein (TXNIP). TXNIP is bound to thiorexodin in resting cells but dissociates in response to high extracellular glucose levels and activates NLRP3. It should be noted 9 that NLRP3, ASC and caspase 1 expression was much higher in bone-marrow derived macrophages than in islet cells[38]. The NLRP3 inflammasome and age-related macular degeneration Age-related macular degeneration (AMD) is a disease that can result in loss of central vision when it progresses to more severe forms such as geographic atrophy (GA). It is characterized by the accumulation of drusen. Drusen is an extracellular deposition of proteins (e.g. complement components, amyloid-β), lipids and advanced glycation end products. In one study, six different proteins present in drusen were tested for their ability to induce IL-1β release from the human retinal pigment cell line ARPE-19. Only N-retinyliddene-N-retinylethanolamine (A2E) triggered IL-1β release. Subsequent experiments show that this release is dependent on the NLRP3 inflammasome and the enzymatic activity of the lysosomal protease cathepsin-B[39]. Surprisingly, amyloid-β (1-42) did not trigger IL-1β release. Amyloid-β is known to result in lysosomal rupture and subsequent NLRP3 inflammasome activation. It should be noted that the authors did not prime the cells to upregulate IL-1β and inflammasome components and this may possibly explain why amyloid-β did not activate inflammasomes in this study. A different study does show NLRP3 inflammasome activation in response to the less toxic amyloid-β (1-40) in rat eyes. This activation was not characterized by cell death in the used model[40]. Another characteristic of AMD is the accumulation of the noncoding retrotransposon RNA sequence Alu. This accumulation of Alu RNA has been implicated in disease progression to GA by a mechanism of NLRP3 inflammasome dependent cell death. Alu RNA results in ROS production and subsequent NLRP3 inflammasome activation and cytotoxicity in retinal pigmented epithelium cells (RPE) in both mice and humans. This pyroptosis-independent cytotoxicity is dependent on activation of MyD88 and IL-18 but not on TLRs[41]. This is an interesting finding since this mechanism may also exist in other celltypes. A more recent study shows that this NLRP3 activation is dependent on the ATP-dependent ion channel P2X7[42]. The RNAse DICER1 was shown to have a protective role against the Alu RNA induced cell death by cleaving Alu RNA. AMD is characterized by loss of DICER1 expression. These findings were confirmed in vivo in RPE cell-specific DICER1 knockout mice and by inhibition of inflammasome components. Additionally, increased caspase-1 activation, released IL-18 and expression of NLRP3, ASC and caspase-1 were observed in the eyes of patients with GA compared to eyes from healthy control[39]. Surprisingly, the observed cell death was independent of pyroptosis. Also RIG-1 activation via a different double stranded RNA species did not result in cell death. I would expect RIG-I inflammasome formation to also result in caspase-1 activation and IL-18 processing. This suggests NLRP3 inflammasome formation activates additional signals that are required for MyD88 and IL-18 dependent cell death as well. Alternatively, additional exogenous signals may be required for RIG-I and/or AIM-2 inflammasome activation and subsequent cell death. A different study reports a protective effect of IL-18 on the development of wet AMD in a mouse model of laser induced choroidal neovascularization (CNV). IL-18 and NLRP3 knockout mice have increased lesions volume compared to wt mice. The amount of IL-1β released by ARPE-19 cells was also compared to the amount of IL-1β released by PBMCs in response to ATP after LPS priming. PMBCs released ~25 fold higher levels ofIL-1β compared to ARPE-19 cells and the authors conclude from this that infiltrated myeloid cells are likely to be mainly responsible for IL-1β and IL-18 release in the retina[43]. Collectively, these studies strongly suggest the involvement of NLRP3 inflammasomes in nonimmune cells during AMD. Whether this has a detrimental or protective effect on disease progression is less clear. Doyle et al. 2012. Observed a protective effect of IL-18[43] whereas Tarallo et al. 2012 observe a detrimental effect on AMD progression[41]. Both studies use a different model and readout 10 for the effect on disease, which may help to explain the observed differences. Whether IL-18 is detrimental or protective for disease progression could also be dependent on the stage of the AMD progression. More studies are required to determine the effects of the observed inflammasome activation and to better characterize the relative role of non-immune cells in AMD compared to immune cells. Ischemia and reperfusion injury A role for inflammasomes has also been implicated in ischemia/reperfusion (I/R) injury. Kawaguchi et al. observed less I/R injury in ASC knockout mice compared to wildtype mice after a 30 minute occlusion of the left anterior descending artery (a branch of the coronary artery) followed by 48 hours of reperfusion[37]. Heart function was also improved and less scarring was observed in ASC knockout mice compared to wt mice at 7 and 14 days after induced myocardial infarction. Similar results were observed with caspase-1 knockout mice. Adoptive bone marrow experiments with ASC knockout mice show that inflammasomes in both immune and non-immune cells are involved in I/R injury. In vitro experiments indicate that cardiac fibroblasts but not cardiomyocytes display inflammasome activation during I/R injury. IL-1β release from cardiac fibroblast but not from cardiomyocytes could be induced with various stimuli (hypoxia/reoxygenation, the K+ ionophore nigericin)[37]. The inflammasome type was not experimentally confirmed, but considering the stimuli they used, the inhibition of IL-1β release by extracellular K+ and the observed production of ROS suggest that the NLRP3 inflammasome is likely to be involved. A more recent study confirms that the NLRP3 inflammasome is involved here. Additionally, they exclude a role in inflammasome activation for infiltrated immune cells in the cardiac tissue by using an ex vivo langendorf model [44]. Contrary to Kawaguchi et al.[37], Mezzaroma et al. did observe (NLRP3) inflammasome activation in the mouse cardiomyocyte cell line HL-1 in response to ATP with LPS priming and hypoxia[45]. This also resulted in cell death dependent on caspase-1 activity since an inhibitor of caspase-1 reduced cell death[45]. This cell death is likely to be pyroptosis. The seemingly conflicting results on the role of cardiac myocytes may be explained by the use of a different concentration of ATP. Kawaguchi et al. use an ATP concentration of 10uM[37] whereas Mezzaroma et al. use between 1 and 5 mM[45]. The stimuli that did trigger inflammasome activation in cardiac fibroblasts but not in cardiomyocytes were not tested by Mezzaroma et al. Sandanger et al. also observe inflammasome activation in primary mouse cardiac fibroblasts in response to ATP at a concentration of 3mM[44] further indicating the concentration may have been too low. Alternatively the observed differences may be caused by characteristics of the HL-1 cell line and primary cardiomyocytes. Atherosclerosis One known promoter of atherosclerosis development is oscillatory shear flow. It has been shown in vitro that oscillatory shear flow upregulates the expression of Sterol regulatory element-binding protein 2 (SREBP2) which indirectly activates the NLRP3 inflammasome. SREBP2 expression results in increased NADPH oxidase expression, which in turn results in ROS production which triggers subsequent NLRP3 inflammasome-dependent IL-1β processing. This study also suggests this occurs in vivo, since they observed increased activated caspase-1 and processed IL-1β in the aortic arch in mice (where higher oscillatory shear flow occurs due to the bend and branches) compared to the thoracic aorta[46]. This observed role of SREBP seems to contradict a previous study where SREBP1 and 2were found to be activated by inflammasome activation resulting in membrane biogenesis[16]. This could indicate a celltype specific role for SREBP2. Alternatively, if both functions apply to the same celltypes, it could be a positive feedback loop. A different study observes increased IL-1β release and caspase-1 activity in vitro in primary rat vascular smooth muscle cells that were calcified with β11 glycerophosphate. This increase is dependent on the NLRP3 inflammasome since knockdown of NLRP3 reduced IL-1β release and caspase-1 activation. They also observed increased caspase-1 activity in calcified human arteries compared to non-calcified arteries in the same patient, suggesting that calcification of arteries may also activate inflammasomes in humans[47]. These studies suggest that the NLRP3 inflammasome is activated by oscillatory shear flow and vessel calcification. All of the studies on inflammasome activation in non-immune cells in response to self-molecules found a primary role for NLRP3 inflammasomes except in the case of psoriasis where AIM-2 was identified as the molecular sensor. Additionally, all of the activating signals appear to be danger signals. Inflammasome activation appears to result in more severe disease progression in diabetes, hyperhomocysteinemia, ischemia/reperfusion injury and possibly atherosclerosis. Most studies on inflammasomes in AMD also suggest detrimental effect of inflammasome activation on AMD progression. However, Doyle et al. 2012 report a protective effect[43]. 5. Response to non-biological substances and stimuli Inflammasome activation in non-immune cells also occurs in response to wide range of substances, such as asbestos, urban particle matter, cigarette smoke and titanium dioxide. Many of these substances can be encountered in everyday life. Beside chemical substances, exposure to UVB also results in inflammasome activation in non-immune cells. Urban particle matter results in NLRP3 and caspase-1 dependent release of IL-1β in human airway epithelial cells[48]. Exposure of wildtype mice to urban particle matter also resulted in increased levels of IL-1β in bronchoalveolar lavage fluid compared to unexposed mice. This effect was not observed in NLRP3 knockout mice. Whether the observed increase in IL-1β is caused by immune cells, pulmonary epithelial cells or both has not been determined. The mice displayed only very few clinical symptoms of pulmonary inflammation[48]. Another study shows NLRP3 dependent inflammasome activation and IL-1β release in a human bronchial epithelial cell line (BEAS-2B) and primary human bronchial epithelial cells in response to silica particles. A much higher (22 fold) IL-1β release was observed in THP-1 cells compared to BEAS-2B cells in response to the same concentration of silica particles[49]. Titanium dioxide nanoparticles, which are frequently used as a white pigment, also trigger inflammasome activation. However, the role of non-immune cells is not fully characterized. Exposure of cultured human keratinocytes and leukemic monocyte THP-1 cells to titanium dioxide nanoparticles resulted caspase-1 activation and IL-1β processing. This response was shown to be dependent on NLRP3 in mouse and human bone marrow-derived macrophages. However, prolonged exposure of mouse skin to titanium dioxide nanoparticles did not result in clinical signs of inflammation. They did observe epidermal thickening which may indicate increased proliferation. Pulmonary exposure did result in inflammation, but whether non-immune cells also play a role here was not determined[50]. Cigarette smoke has been shown to trigger IL-1β release in a cultured human bronchial epithelial cell line (HBE-14o). This release was blocked by a caspase-1or a P2x7 inhibitor and also by a ROS scavenger. It was not determined whether ASC and which molecular sensor(s) are involved. Considering that inhibition of P2x7 or a ROS scavenger inhibited IL-1β release, it appears likely that the NLRP3 inflammasome is involved[51]. Cultured human mesothelial cells exposed to crocidolite (a type of asbestos) or the mineral erionite, which has similar properties to asbestos in some regards, show increased caspase-1 activity and IL-1β and IL-18 release compared to control cells. This increase was inhibited by NLRP3 knockdown. Treatment with the IL-1 receptor antagonist Anakinra reduced IL-1β release, suggesting autocrine stimulation[52]. 12 Non chemical stimulation in the form of UVB radiation has also been reported to result in inflammasome activation in keratinocytes. Stimulation of primary human keratinocytes with UVB results in IL-1β release but not the release of IL-18. The IL-1β release was inhibited by the knockdown of caspase-1, ASC, NLRP1 or NLRP3 showing the involvement of the NLRP1 and NLRP3 inflammasome. The IL-1β release was also inhibited by a calcium chelator, indicating the dependence on a calcium influx. Additionally, a reduced number of neutrophils were detected in UVB irradiated skin of caspase-1 knockout mice compared to wildtype mice, suggesting a role for inflammasomes in recruiting them to the site of inflammation[53]. A more recent study also determined that UVB-induced IL-1β release from keratinocytes is dependent on activation of human caspase-4 but not caspase-5. Caspase-4 and -5 are likely to be orthologs of mouse caspase-11[54]. A number of studies report on NLRP3 inflammasome activation in lung epithelial cells and on NLRP1 and NLRP3 inflammasome activation in keratinocytes. However, whether inflammasome activation in response to these stimuli has beneficial or pathological consequences is not well characterized. In vivo studies will be required to determine this. It should also be noted that keratinocytes in the skin are normally not directly in contact with most external stimuli, since they are covered by layers of dead keratinocytes. This may also explain why exposure of mouse skin to titanium dioxide nanoparticles did not result in inflammation whereas pulmonary exposure in mice did[50].It is interesting to note that all of the above mentioned stimuli are known to or have been implicated to be carcinogenic. Since inflammasomes are known to be involved in carcinogenesis (see below) there could be a mechanistic link here. 6. Inflammasomes in wound healing, tumor development and pain sensitivity Effects of inflammasome activation have also been reported in the development in tumors and cell proliferation in skin and the intestine. Most of these studies report a protective effect of inflammasomes against tumor formation. However, some studies report that inflammasomes may promote tumor development. One study shows an inflammasome-independent role for ASC in keratinocytes in suppressing tumor formation. A number of studies also suggest a role for inflammasome activation in promoting or repressing cell proliferation and wound healing. Wound healing and tumors Caspase-1 knockout mice have an increased number and severity of tumors in the colon in a model of chemically induced tumorigenesis compared to wt mice. Increased proliferation of colon epithelial cells and reduced cell death in tumors of caspase-1 knockout mice was observed compared to wildtype control mice. The increased number of tumors and tumor severity was observed in NLRC4 knockout mice but not NLRP3 mice. This suggests a role for NLRC4 but not NLRP3 in this model[55]. The underlying activating signal for NLRC4 inflammasome activation has not been elucidated. The authors hypothesize based on the well-established activation of NLRC4 by Gramnegative bacteria that altered microbiota in the colon may play a role. Although the increased proliferation was observed in colon epithelial cells, a role for immune cells in the observed knockout phenotypes cannot be excluded. The observed decrease in cell death in tumor cells may possibly be caused by deficient pyroptosis. In another study, reduced colon healing was observed in NLRP6 knockout mice after causing biopsy injury. There was also an increase in the number of tumors in NLRP6, ASC and caspase-1 knockout mice in a model of chemically induced tumorigenesis. Adoptive transfer of bone-marrow of caspase-1 knockout or wildtype into caspase-1 knockout or wildtype mice reveal that caspase-1 is required in both hematopoietic and non-hematopoietic cells to acquire the tumor phenotype of caspase-1 knockout mice, since neither of the partial knockouts was sufficient to acquire the 13 phenotype[56]. This study suggests that inflammasome activation in non-immune cells together with inflammasomes activation in immune cells can promote tumorigenesis and wound healing. A mechanistic link between inflammasome activation and the observed phenotypes has not been found yet. Another study reports a protective effect of inflammasome activation on tumorigenesis but concludes this is primary caused by inflammasome activation in immune cells. Experiments with adoptive transfer of bone marrow of NLRP3 knockout and wildtype mice show that NLRP3 inflammasome activation in hematopoietic cells is primarily responsible for the observed protective effect of the inflammasome on tumorigenesis[57]. These results do not exclude the possibility that another molecular sensor may be activated in non-hematopoietic cells in this model. In a different study, IL-18 has been shown to promote proliferation of intestinal epithelial cells in mice by indirectly increasing IL-22 expression[58]. IL-18 downregulates the expression of IL-22bp. IL-22bp is expressed mostly in hematopoietic cells and inhibits IL-22. IL-22 promotes proliferation of intestinal epithelial and tumor cells. They also show that the expression of IL-22bp is dependent on NLRP3 and NLRP6 but not NLRC4. Therefore, inflammasome activation results in IL-18 release, which reduces IL-22bp expression in hematopoietic cells, which in turn abolishes inhibition of IL-22 which subsequently results in increased proliferation of intestinal epithelial and tumor cells[58]. This study provides a mechanism on how inflammasome activation may promote tumorigenesis and wound healing in the intestine. This study also reports that the microbiota affects the downregulation of IL22bp[58]. Whether the inflammasome activation occurs in epithelial or hematopoietic cells is not known in this study. A role for inflammasomes in promoting wound healing in the skin has also been implicated by affecting the release of processed IL-1α[12]. One study shows an inflammasome-independent role for ASC as a tumor suppressor in keratinocytes. At the same time they show an inflammasome-dependent tumor promoting role in immune cells. Reduced skin tumor formation after treatment with carcinogenic substances in IL-1R and caspase-1 knockout mice was observed compared to the wt control. However, this was not observed for ASC knockout mice. They then used myeloid and keratinocyte specific ASC knockout mice to try to explain these seemingly conflicting results. Myeloid ASC knockout mice show fewer tumors whereas keratinocyte ASC knockout mice have more tumors compared to wildtype control mice. A reduction in processed IL-1β was observed in tumors of myeloid ASC knockout mice compared to the control whereas no difference between the wt control was observed for keratinocyte ASC knockout mice tumors. This suggests that infiltrating myeloid cells are the primary source of processed IL-1β rather than keratinocytes in this model of skin tumors. In vitro experiments revealed increased proliferation in keratinocytes from ASC knockout mice compared to the control whereas caspase-1 and NLRP3 knockout keratinocytes did not. They also observe decreased ASC expression in tumors compared to healthy skin. Additionally, they observe reduced P53 phosphorylation and downstream p21 and Notch1 expression in keratinocytes from knockout mice compared to keratinocytes from healthy control mice. Together, these data suggest an inflammasome independent role for ASC as a tumor suppressor in keratinocytes which may be mediated by regulating P53 activity[6]. Inflammasomes do not appear to play a role in keratinocytes in this study despite the many studies that do observe inflammasome activation and IL-1β processing in keratinocytes. Infiltrating myeloid cells on the other hand do show inflammasome-dependent IL-1β release in these tumors. This observed cell type specific inflammasome activation could be mediated by additional control factors that are differentially expressed in different cell types. Pain sensitivity Inflammasome dependent processing of IL-1β and IL-18 in keratinocytes has also been implicated in increasing pain sensitivity. In a rat model of tibia fracture, increased NLRP1 and caspase-1 expression 14 was observed in keratinocytes in the skin around the fracture and also elevated levels of released IL1β and IL-18 compared to non-fractured limbs. Injection of IL-18 and IL-1β in a limb also increased the sensitivity to mechanical pain induction. The inhibition of caspase-1 decreases the enhanced pain sensation in a limb that has previously been fractured compared to a non-fractured limb[59]. They did not determine the activating signal for inflammasomes or confirm that NLRP1 inflammasomes process IL-1β and IL-18. Taken together, these studies show that inflammasomes in non-immune cells as well as in immune cells are likely to be involved in tumor formation and wound healing in the skin and the intestine. Beside these processes, inflammasome activation in keratinocytes has also been implicated in increasing pain sensitivity. Whether inflammasome activation stimulates or diminishes wound healing and tumor formation is less clear. Most studies report a stimulatory effect of inflammasomes on proliferation. However, Hu et al. 2010 report a decline in proliferation of colon epithelium[55]. Most studies show a decline in tumor formation when inflammasomes are still functional. However, Huber et al. 2012 report a stimulating effect of inflammasomes in either immune and/or non-immune cells on tumorigenesis[58] and Drexler et al. 2012 also report a stimulating effect of inflammasomes in immune cells on tumorigenesis (but an inflammasome-independent protective role for ASC against tumor formation in non-immune cells)[6]. All the studies that report a protective effect of inflammasome activation on tumor formation conclude that this is caused by inflammasomes in nonimmune cells. This could suggest that inflammasome activation in non-immune cells has a protective effect against tumorigenesis whereas inflammasome activation in immune cells promotes tumor formation. Additional regulatory mechanisms may also be involved which may help explain the different results in these studies. Inflammasome activation may also have a protective or stimulating effect on tumors depending on the stage of tumor development. A role for inflammasomes in affecting microbiota, which in turn may affect tumor development has also been suggested but no yet investigated. 15 Overview of inflammasome types in non-immune cells Cell type(s) species In vivo or in vitro Inflammasome types Activating signals Possible effect of inflammasome activation references mouse In vivo NLRP3 Microbiota Changes in microbiota, less sever NASH symptoms [21] Mouse in vivo NLP6 Microbiota Less severe colitis [22] Colon epithelial cells Mouse In vivo NLRC4 reduced proliferation [55] Colon epithelial cells Mouse In vivo NLRP6 increased proliferation [56] intestinal epithelial and goblet cells Mouse In vivo NLRP6 Increased mucus secretion [26] Caco-2 Human In vitro NLRP3 Y. enterocolitica IL-18 release (but no IL-1β) [23] Human In vitro NLRP3 Chlamydia trachomatis Increased c. trachomatitis infectivity in vitro [17] Primary lung epithelial cells human In vitro NLP3, RIG-I Influenza A Lung epithelial cells Mouse In vivo NLRP3 OVA Inflammation [60] HBE-14o Human In vitro NLRP3 (?) Cigarette smoke Inflammation [51] Airway epithelial cells Human In vitro NLRP3 Urban particle matter Inflammation [48] BEAS-2B and primary human bronchial epithelial cells Human In vitro NLRP3 Silica particles Suggestive for effect in silicosis [49] Mesothelialial epithelium cells Human In vitro NLRP3 Asbestos, erionite inflammation [52] Adipocytes Mouse In vivo NLRP3 sucrose increased glucose sensitivity, decreased insulin sensitivity, possible role diabetes [35] Primary adipocytes Human In vitro Sucrose Possible role diabetes [61] Keratinocytes Mouse In vitro NLRP3 (?) UVB, SDS, DNFB Possibly increased inflammation early phase of contact hypersensitivity [28] Primary human keratinocytes Human In vitro NLRP3 Der p 1 Possible role in atopic dermatitis [30] Intestinal tract Non-hepatic non-hematopoietic cells(presumably intestinal epithelial cells) Non-hematopoietic cells (presumably intestinal epithelial cells) Cervix HeLa cells Lung [4] Adipose tissue Skin 16 Primary human and mouse keratinocytes Human and mouse In vitro ? DNFB Keratinocytes Human In vitro AIM-2 (?) Double stranded DNA Melittin, mitochondrial and nuclear DNA DNA localized in cytosol Possible stimulatory role in atopic dermatitis and increased Th2 skewing [29] [62] Inflammation bee sting, possible role hypersensitivity [31] Possible role in psoriasis [34] Increased pain sensitivity [59] Keratinocytes Human In vitro AIM-2 Keratinocytes Human In vivo AIM-2 Keratinocytes Rat In vivo NLRP1 (?) Keratinocytes Human In vitro NLRP3 (?) Titanium dioxide nanoparticles inflammation [50] Keratinocytes Human In vitro NLRP3, NLRP1 UVB Neutrophil influx to site of inflammation [53] Mouse In vivo NLRP3 Homocysteine Hyperhomocysteinemia progression [32] ARPE-19 Human In vitro NLRP3 A2E, ATP AMD progression [39] RPE cells Human and Mouse In vivo NLRP3 Alu RNA AMD progression [41][42] ARPE-19 Human In vitro NLRP3 ATP Protects against AMD [43] cardiac fibroblasts Mouse In vivo and in vitro ? (NLRP3?) Nigericin, hypoxia Ischemia/reperfusion injury [37] HL-1 Mouse In vitro NLRP3 ATP Cell death [45] Cardiac myocytes Mouse NLRP3 ATP Ischemia/reperfusion injury [44] Endothelial cells Human and mouse In vitro and ex vivo In vivo and in vitro NLRP3 Oscillatory shear flow Possible role in atherosclerosis [46] Vascular smooth muscle cells Rat in vitro NLRP3 Calcium phosphate deposits Possible role in atherosclerosis [47] endothelial telomerase-immortalized human umbilical cells Human In vitro IFI16 KSHV genome Inflammation induced by pathogen [10] Islet cells Mouse In vitro NLRP3 Sucrose Possible role in diabetes [38] Kidney Podocytes Eye Heart and blood vessels Other Table 1. Overview of inflammasome types, stimulatory signals and effects of inflammasome activation in non-immune cells. A question mark indicates that the inflammasome type has not been fully confirmed. The in vitro/in vivo indicates whether the inflammasomes in non-immune cells were specifically identified in non-immune cells in vivo or in vitro. 17 7. Discussion Ample evidence exists for a role of inflammasomes in non-immune cells. Inflammasome activation in non-immune cells have been implicated as a response against various stimuli including pathogens, the microbiota, metabolites, self-proteins and self RNA species, hyper immune responses against foreign substances (biological and non-biological), wound healing and cancer. Different molecular sensors of inflammasomes have been identified in non-immune cells including NLRP1, NLRP3, NLRC4, NLRP6, AIM-2, RIG-I and IFI16. NLRP3 is the most frequently involved in these studies. Most of the studies discussed in this review focus on canonical inflammasomes and interleukin production. Caspase-11-dependent inflammasome activation, caspase-8 dependent IL-1β processing, the role of IL-1α as an IL-1R ligand and alternative inflammasome functions are often not taken into consideration. Besides upregulation of inflammasome components (priming), additional regulatory mechanisms of inflammasome activation are often ignored as well. Many studies have inconsistent results regarding the in vivo effect of inflammasome activation on disease. Investigation of additional mechanisms involved in inflammasome activation may help to explain these discrepancies. The inconsistent results may also indicate that inflammasome activation can be beneficial or detrimental depending on the situation or that the right balance in inflammasome activation is required. The same types of inflammasomes are present in immune and non-immune cells. Inflammasome activation in non-immune cells also results in the processing of pro-interleukins as observed in immune cells. Studies that compare the amount of IL-1β released by immune cells to non-immune cells for the same number of cells in vitro[24][49][43] or for the total contribution in an organ or tissue in vivo[25][6] conclude that immune cells release more processed IL-1β. Since many epithelial cells can be continuously exposed to inflammasome stimulating substances, a lower sensitivity to these stimuli may prevent continues inflammasome activation. Despite the lower pro-interleukin processing of non-immune cells, many studies see potent effects of inflammasome activation in nonimmune cells on disease. This suggests that alternative functions of inflammasome activation may mediate these effects. Another possibility is that inflammasome activation in non-immune cells and IL-1β release result in recruitment of immune cells which then further amplify inflammation. Many epithelial tissues have been shown to express functional inflammasomes. Epithelial cells are likely to be the first cells exposed to pathogens and foreign molecules and are therefore likely candidates to initiate inflammation. One study also observed a reduced influx of neutrophils to UVB irradiated skin area in keratinocyte specific caspase-1 knockout mice compared to wildtype mice[53], which supports this theory. Experiments with cultured mesothelial epithelium cells also suggest a role for IL-1β in autocrine signaling to further amplify inflammation[52]. A large number of exogenous stimuli that are frequently encountered in daily life as well selfproteins and metabolites have been shown to be able to trigger inflammasomes in non-immune cells. Many of these signals could be characterized as danger signals. If the primary function of inflammasome activation is to detect pathogens and to initiate an immune response, it seems unbeneficial to do so without detecting a pathogen-specific signal as well when considering the potential damage of inflammation. One explanation could be that it’s an evolutionary tradeoff where the risk of not detecting a pathogen early is greater than the damage caused by occasional accidental inflammasome activation. Alternatively, it could indicate that many of these stimuli do not accidentally activate inflammasomes but instead function to activate alternative functions of inflammasomes, such as proliferation or pyroptosis. All of the non-biological exogenous stimuli discussed in this review are known or very likely to be carcinogenic. Since inflammasome activation has been implicated in increased pain sensitivity[59], inflammasome activation could be a mechanism to cause irritation to trigger a behavioral change to avoid exposure to dangerous substances. Inflammasome activation in response to substances that can cause direct tissue damage may be 18 beneficial to initiate tissue repair and prepare the immune systems for any pathogens that may now be able to pass over the epithelial barrier. Effects of inflammasome activation in non-immune cells on many diseases have frequently been observed. This shows that inflammasome activation in non-immune cells has biological relevance in vivo. Many studies report conflicting results on whether inflammasome activation is beneficial or detrimental in diseases. This indicates a more complex situation then our current understanding. More research is required to determine the functions and effects of inflammasome activation in non-immune cells as well as to better understand non-canonical inflammasomes. Also the relative role of inflammasome activation in non-immune cells compared to immune cells as well as their interplay is currently still poorly understood. A singular role for inflammasomes in immunity does not seem likely in the light of these studies. References [1] E. Latz, T. S. Xiao, and A. Stutz, “Activation and regulation of the inflammasomes.,” Nature reviews. Immunology, vol. 13, no. 6, pp. 397–411, Jun. 2013. [2] K. Schroder and J. Tschopp, “The inflammasomes.,” Cell, vol. 140, no. 6, pp. 821–32, Mar. 2010. [3] I. Sastalla, D. Crown, S. L. Masters, A. McKenzie, S. H. Leppla, and M. Moayeri, “Transcriptional analysis of the three Nlrp1 paralogs in mice.,” BMC genomics, vol. 14, no. 1, p. 188, Jan. 2013. [4] J. Pothlichet, I. Meunier, B. K. Davis, J. P.-Y. Ting, E. Skamene, V. von Messling, and S. M. Vidal, “Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells.,” PLoS pathogens, vol. 9, no. 4, p. e1003256, Apr. 2013. [5] N. B. Bryan, A. Dorfleutner, S. J. Kramer, C. Yun, Y. Rojanasakul, and C. Stehlik, “Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes.,” Journal of inflammation (London, England), vol. 7, p. 23, Jan. 2010. [6] S. K. Drexler, L. Bonsignore, M. Masin, A. Tardivel, R. Jackstadt, and H. Hermeking, “Tissue-speci fi c opposing functions of the in fl ammasome adaptor ASC in the regulation of epithelial skin carcinogenesis,” 2012. [7] E. Viganò and A. Mortellaro, “Caspase-11: The driving factor for noncanonical inflammasomes.,” European journal of immunology, vol. 43, no. 9, pp. 2240–5, Sep. 2013. [8] P. Broz and D. M. Monack, “Noncanonical inflammasomes: caspase-11 activation and effector mechanisms.,” PLoS pathogens, vol. 9, no. 2, p. e1003144, Feb. 2013. [9] G. Sollberger, G. E. Strittmatter, M. Garstkiewicz, J. Sand, and H.-D. Beer, “Caspase-1: The inflammasome and beyond.,” Innate immunity, May 2013. [10] V. V. Singh, N. Kerur, V. Bottero, S. Dutta, S. Chakraborty, M. A. Ansari, N. Paudel, L. Chikoti, and B. Chandran, “Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes.,” Journal of virology, vol. 87, no. 8, pp. 4417–31, Apr. 2013. [11] A. S. Yazdi and S. K. Drexler, “Regulation of interleukin 1α secretion by inflammasomes.,” Annals of the rheumatic diseases, vol. 72 Suppl 2, pp. ii96–9, Apr. 2013. [12] P. Lee, D.-J. Lee, C. Chan, S.-W. Chen, I. Ch’en, and C. Jamora, “Dynamic expression of epidermal caspase 8 simulates a wound healing response.,” Nature, vol. 458, no. 7237, pp. 519–23, Mar. 2009. 19 [13] E. a Miao, J. V Rajan, and A. Aderem, “Caspase-1-induced pyroptotic cell death.,” Immunological reviews, vol. 243, no. 1, pp. 206–14, Sep. 2011. [14] W. Shao, G. Yeretssian, K. Doiron, S. N. Hussain, and M. Saleh, “The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock.,” The Journal of biological chemistry, vol. 282, no. 50, pp. 36321–9, Dec. 2007. [15] L. Gurcel, L. Abrami, S. Girardin, J. Tschopp, and F. G. van der Goot, “Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival.,” Cell, vol. 126, no. 6, pp. 1135–45, Sep. 2006. [16] S.-S. Im, L. Yousef, C. Blaschitz, J. Z. Liu, R. a Edwards, S. G. Young, M. Raffatellu, and T. F. Osborne, “Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a.,” Cell metabolism, vol. 13, no. 5, pp. 540–9, May 2011. [17] A. a Abdul-Sater, E. Koo, G. Häcker, and D. M. Ojcius, “Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen Chlamydia trachomatis.,” The Journal of biological chemistry, vol. 284, no. 39, pp. 26789–96, Sep. 2009. [18] R. Muñoz-Planillo, P. Kuffa, G. Martínez-Colón, B. L. Smith, T. M. Rajendiran, and G. Núñez, “K + efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter.,” Immunity, vol. 38, no. 6, pp. 1142–53, Jun. 2013. [19] W. A. Tseng, T. Thein, K. Kinnunen, K. Lashkari, M. S. Gregory, P. a D’Amore, and B. R. Ksander, “NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration.,” Investigative ophthalmology & visual science, vol. 54, no. 1, pp. 110– 20, Jan. 2013. [20] J. Henao-Mejia, E. Elinav, T. Strowig, and R. a Flavell, “Inflammasomes: far beyond inflammation.,” Nature immunology, vol. 13, no. 4, pp. 321–4, Apr. 2012. [21] J. Henao-Mejia, E. Elinav, C. Jin, L. Hao, W. Z. Mehal, T. Strowig, C. a Thaiss, A. L. Kau, S. C. Eisenbarth, M. J. Jurczak, J.-P. Camporez, G. I. Shulman, J. I. Gordon, H. M. Hoffman, and R. a Flavell, “Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity.,” Nature, vol. 482, no. 7384, pp. 179–85, Feb. 2012. [22] E. Elinav, T. Strowig, A. L. Kau, J. Henao-Mejia, C. a Thaiss, C. J. Booth, D. R. Peaper, J. Bertin, S. C. Eisenbarth, J. I. Gordon, and R. a Flavell, “NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis.,” Cell, vol. 145, no. 5, pp. 745–57, May 2011. [23] J. Thinwa, J. a. Segovia, S. Bose, and P. H. Dube, “Integrin-Mediated First Signal for Inflammasome Activation in Intestinal Epithelial Cells,” The Journal of Immunology, Jun. 2014. [24] A. Tang, A. Sharma, R. Jen, A. F. Hirschfeld, M. a Chilvers, P. M. Lavoie, and S. E. Turvey, “Inflammasome-mediated IL-1β production in humans with cystic fibrosis.,” PloS one, vol. 7, no. 5, p. e37689, Jan. 2012. [25] T. S. Cohen and A. S. Prince, “Activation of inflammasome signaling mediates pathology of acute P . aeruginosa pneumonia,” vol. 123, no. 4, 2013. [26] M. Wlodarska, C. a Thaiss, R. Nowarski, J. Henao-Mejia, J.-P. Zhang, E. M. Brown, G. Frankel, M. Levy, M. N. Katz, W. M. Philbrick, E. Elinav, B. B. Finlay, and R. a Flavell, “NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion.,” Cell, vol. 156, no. 5, pp. 1045–59, Feb. 2014. [27] L. Feldmeyer, S. Werner, L. E. French, and H.-D. Beer, “Interleukin-1, inflammasomes and the skin.,” European journal of cell biology, vol. 89, no. 9, pp. 638–44, Sep. 2010. 20 [28] H. Watanabe, O. Gaide, V. Pétrilli, F. Martinon, E. Contassot, S. Roques, J. a Kummer, J. Tschopp, and L. E. French, “Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity.,” The Journal of investigative dermatology, vol. 127, no. 8, pp. 1956–63, Aug. 2007. [29] S. Schuepbach-Mallepell, V. Philippe, M.-C. Brüggen, H. Watanabe, S. Roques, C. Baldeschi, and O. Gaide, “Antagonistic effect of the inflammasome on thymic stromal lymphopoietin expression in the skin.,” The Journal of allergy and clinical immunology, pp. 1–10, Aug. 2013. [30] X. Dai, K. Sayama, M. Tohyama, Y. Shirakata, Y. Hanakawa, S. Tokumaru, L. Yang, S. Hirakawa, and K. Hashimoto, “Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes.,” The Journal of allergy and clinical immunology, vol. 127, no. 3, pp. 806–14.e1–4, Mar. 2011. [31] Y. Dombrowski, M. Peric, S. Koglin, N. Kaymakanov, V. Schmezer, M. Reinholz, T. Ruzicka, and J. Schauber, “Honey bee (Apis mellifera) venom induces AIM2 inflammasome activation in human keratinocytes.,” Allergy, vol. 67, no. 11, pp. 1400–7, Nov. 2012. [32] C. Zhang, K. M. Boini, M. Xia, J. M. Abais, X. Li, Q. Liu, and P.-L. Li, “Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia.,” Hypertension, vol. 60, no. 1, pp. 154–62, Jul. 2012. [33] J. M. Abais, C. Zhang, M. Xia, Q. Liu, T. W. B. Gehr, K. M. Boini, and P.-L. Li, “NADPH oxidasemediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia.,” Antioxidants & redox signaling, vol. 18, no. 13, pp. 1537–48, May 2013. [34] Y. Dombrowski, M. Peric, S. Koglin, C. Kammerbauer, C. Göss, D. Anz, M. Simanski, R. Gläser, J. Harder, V. Hornung, R. L. Gallo, T. Ruzicka, R. Besch, and J. Schauber, “Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions.,” Science translational medicine, vol. 3, no. 82, p. 82ra38, May 2011. [35] R. Stienstra, L. a B. Joosten, T. Koenen, B. van Tits, J. a van Diepen, S. a a van den Berg, P. C. N. Rensen, P. J. Voshol, G. Fantuzzi, A. Hijmans, S. Kersten, M. Müller, W. B. van den Berg, N. van Rooijen, M. Wabitsch, B.-J. Kullberg, J. W. M. van der Meer, T. Kanneganti, C. J. Tack, and M. G. Netea, “The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity.,” Cell metabolism, vol. 12, no. 6, pp. 593–605, Dec. 2010. [36] B. Vandanmagsar, Y.-H. Youm, A. Ravussin, J. E. Galgani, K. Stadler, R. L. Mynatt, E. Ravussin, J. M. Stephens, and V. D. Dixit, “The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance.,” Nature medicine, vol. 17, no. 2, pp. 179–88, Feb. 2011. [37] M. Kawaguchi, M. Takahashi, T. Hata, Y. Kashima, F. Usui, H. Morimoto, A. Izawa, Y. Takahashi, J. Masumoto, J. Koyama, M. Hongo, T. Noda, J. Nakayama, J. Sagara, S. Taniguchi, and U. Ikeda, “Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury.,” Circulation, vol. 123, no. 6, pp. 594–604, Feb. 2011. [38] R. Zhou, A. Tardivel, B. Thorens, I. Choi, and J. Tschopp, “Thioredoxin-interacting protein links oxidative stress to inflammasome activation.,” Nature immunology, vol. 11, no. 2, pp. 136–40, Feb. 2010. [39] O. a Anderson, A. Finkelstein, and D. T. Shima, “A2E induces IL-1ß production in retinal pigment epithelial cells via the NLRP3 inflammasome.,” PloS one, vol. 8, no. 6, p. e67263, Jan. 2013. [40] R. T. Liu, J. Gao, S. Cao, N. Sandhu, J. Z. Cui, C. L. Chou, E. Fang, and J. a Matsubara, “Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: implications for inflammasome activation in age-related macular degeneration.,” Investigative ophthalmology & visual science, vol. 54, no. 3, pp. 2225–37, Mar. 2013. [41] V. Tarallo, Y. Hirano, B. D. Gelfand, S. Dridi, N. Kerur, Y. Kim, W. G. Cho, H. Kaneko, B. J. Fowler, S. Bogdanovich, R. J. C. Albuquerque, W. W. Hauswirth, V. a Chiodo, J. F. Kugel, J. a Goodrich, S. L. 21 Ponicsan, G. Chaudhuri, M. P. Murphy, J. L. Dunaief, B. K. Ambati, Y. Ogura, J. W. Yoo, D. Lee, P. Provost, D. R. Hinton, G. Núñez, J. Z. Baffi, M. E. Kleinman, and J. Ambati, “DICER1 loss and Alu RNA induce agerelated macular degeneration via the NLRP3 inflammasome and MyD88.,” Cell, vol. 149, no. 4, pp. 847–59, May 2012. [42] N. Kerur, Y. Hirano, V. Tarallo, B. Fowler, A. Bastos-Carvalho, T. Yasuma, R. Yasuma, Y. Kim, D. R. Hinton, C. J. Kirschning, B. Gelfand, and J. Ambati, “TLR independent and P2X7 dependent signaling regulates Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy.,” Investigative ophthalmology & visual science, pp. 3–9, Oct. 2013. [43] S. L. Doyle, M. Campbell, E. Ozaki, R. G. Salomon, A. Mori, P. F. Kenna, G. J. Farrar, A.-S. Kiang, M. M. Humphries, E. C. Lavelle, L. a J. O’Neill, J. G. Hollyfield, and P. Humphries, “NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components.,” Nature medicine, vol. 18, no. 5, pp. 791–8, May 2012. [44] Ø. Sandanger, T. Ranheim, L. E. Vinge, M. Bliksøen, K. Alfsnes, A. V Finsen, C. P. Dahl, E. T. Askevold, G. Florholmen, G. Christensen, K. a Fitzgerald, E. Lien, G. Valen, T. Espevik, P. Aukrust, and A. Yndestad, “The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury.,” Cardiovascular research, vol. 99, no. 1, pp. 164–74, Jul. 2013. [45] E. Mezzaroma, S. Toldo, D. Farkas, I. M. Seropian, B. W. Van Tassell, F. N. Salloum, H. R. Kannan, A. C. Menna, N. F. Voelkel, and A. Abbate, “The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse.,” Proceedings of the National Academy of Sciences of the United States of America, vol. 108, no. 49, pp. 19725–30, Dec. 2011. [46] H. Xiao, M. Lu, T. Y. Lin, Z. Chen, G. Chen, W.-C. Wang, T. Marin, T.-P. Shentu, L. Wen, B. Gongol, W. Sun, X. Liang, J. Chen, H.-D. Huang, J. H. F. Pedra, D. a Johnson, and J. Y. J. Shyy, “Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility.,” Circulation, vol. 128, no. 6, pp. 632–42, Aug. 2013. [47] C. Wen, X. Yang, Z. Yan, M. Zhao, X. Yue, X. Cheng, Z. Zheng, K. Guan, J. Dou, T. Xu, Y. Zhang, T. Song, C. Wei, and H. Zhong, “Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification.,” International journal of cardiology, vol. 168, no. 3, pp. 2242–7, Oct. 2013. [48] J. a Hirota, S. a Hirota, S. M. Warner, D. Stefanowicz, F. Shaheen, P. L. Beck, J. a Macdonald, T.-L. Hackett, D. D. Sin, S. Van Eeden, and D. a Knight, “The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter.,” The Journal of allergy and clinical immunology, vol. 129, no. 4, pp. 1116–25.e6, Apr. 2012. [49] P. M. Peeters, T. N. Perkins, E. F. M. Wouters, B. T. Mossman, and N. L. Reynaert, “Silica induces NLRP3 inflammasome activation in human lung epithelial cells.,” Particle and fibre toxicology, vol. 10, no. 1, p. 3, Jan. 2013. [50] A. S. Yazdi, G. Guarda, N. Riteau, S. K. Drexler, A. Tardivel, I. Couillin, and J. Tschopp, “Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1α and IL-1β.,” Proceedings of the National Academy of Sciences of the United States of America, vol. 107, no. 45, pp. 19449–54, Nov. 2010. [51] E. Mortaz, P. a J. Henricks, a D. Kraneveld, M. E. Givi, J. Garssen, and G. Folkerts, “Cigarette smoke induces the release of CXCL-8 from human bronchial epithelial cells via TLRs and induction of the inflammasome.,” Biochimica et biophysica acta, vol. 1812, no. 9, pp. 1104–10, Sep. 2011. [52] J. M. Hillegass, J. M. Miller, M. B. MacPherson, C. M. Westbom, M. Sayan, J. K. Thompson, S. L. Macura, T. N. Perkins, S. L. Beuschel, V. Alexeeva, H. I. Pass, C. Steele, B. T. Mossman, and A. Shukla, “Asbestos and erionite prime and activate the NLRP3 inflammasome that stimulates autocrine cytokine release in human mesothelial cells.,” Particle and fibre toxicology, vol. 10, no. 1, p. 39, Jan. 2013. 22 [53] L. Feldmeyer, M. Keller, G. Niklaus, D. Hohl, S. Werner, and H.-D. Beer, “The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes.,” Current biology : CB, vol. 17, no. 13, pp. 1140–5, Jul. 2007. [54] G. Sollberger, G. E. Strittmatter, M. Kistowska, L. E. French, and H.-D. Beer, “Caspase-4 is required for activation of inflammasomes.,” Journal of immunology (Baltimore, Md. : 1950), vol. 188, no. 4, pp. 1992– 2000, Feb. 2012. [55] B. Hu, E. Elinav, S. Huber, C. J. Booth, T. Strowig, C. Jin, S. C. Eisenbarth, and R. a Flavell, “Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4.,” Proceedings of the National Academy of Sciences of the United States of America, vol. 107, no. 50, pp. 21635–40, Dec. 2010. [56] S. Normand, A. Delanoye-Crespin, A. Bressenot, L. Huot, T. Grandjean, L. Peyrin-Biroulet, Y. Lemoine, D. Hot, and M. Chamaillard, “Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury.,” Proceedings of the National Academy of Sciences of the United States of America, vol. 108, no. 23, pp. 9601–6, Jun. 2011. [57] I. C. Allen, E. M. TeKippe, R.-M. T. Woodford, J. M. Uronis, E. K. Holl, A. B. Rogers, H. H. Herfarth, C. Jobin, and J. P.-Y. Ting, “The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer.,” The Journal of experimental medicine, vol. 207, no. 5, pp. 1045–56, May 2010. [58] S. Huber, N. Gagliani, L. a Zenewicz, F. J. Huber, L. Bosurgi, B. Hu, M. Hedl, W. Zhang, W. O’Connor, A. J. Murphy, D. M. Valenzuela, G. D. Yancopoulos, C. J. Booth, J. H. Cho, W. Ouyang, C. Abraham, and R. a Flavell, “IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine.,” Nature, vol. 491, no. 7423, pp. 259–63, Nov. 2012. [59] W.-W. Li, T.-Z. Guo, D. Liang, X. Shi, T. Wei, W. S. Kingery, and J. D. Clark, “The NALP1 inflammasome controls cytokine production and nociception in a rat fracture model of complex regional pain syndrome.,” Pain, vol. 147, no. 1–3, pp. 277–86, Dec. 2009. [60] H. B. Tran, M. D. Lewis, L. W. Tan, S. E. Lester, L. M. Baker, J. Ng, M. a Hamilton-Bruce, C. L. Hill, S. a Koblar, M. Rischmueller, R. E. Ruffin, P. J. Wormald, P. D. Zalewski, and C. J. Lang, “Immunolocalization of NLRP3 Inflammasome in Normal Murine Airway Epithelium and Changes following Induction of Ovalbumin-Induced Airway Inflammation.,” Journal of allergy, vol. 2012, p. 819176, Jan. 2012. [61] M. Lappas, “Activation of inflammasomes in adipose tissue of women with gestational diabetes.,” Molecular and cellular endocrinology, vol. 382, no. 1, pp. 74–83, Sep. 2013. [62] V. Kopfnagel, M. Wittmann, and T. Werfel, “Human keratinocytes express AIM2 and respond to dsDNA with IL-1β secretion.,” Experimental dermatology, vol. 20, no. 12, pp. 1027–9, Dec. 2011. 23