Lab.2.background - Cal State LA
advertisement

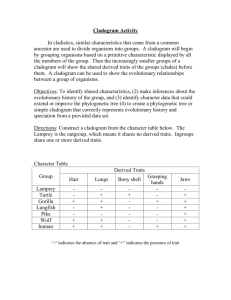
Lab two – Individual adaptations to the environment: leaf convergence in plants Objectives – after completing this laboratory, you should: Clarify your understanding of what an adaptation is. Understand the concept of morphological convergence and its underlying causes. Become familiar with the specialized leaf structures of carnivorous plants. Be familiar with evolutionary tree diagrams, or cladograms, and what they represent Be able to use a cladogram to test hypotheses of convergence and adaptation. Background – When two organisms share a feature, there are really two explanations for how this came to be: One possibility is that the organisms share the feature because they both inherited it from a common ancestor they shared some time in the past. In evolutionary biology, a feature that is shared by two different species through common descent from the same ancestor is known as a homology, and the features in each organism are described as being homologous. However, a second possibility exists: it is also possible that two species share the same feature because they each evolved it independently. In this case, if you were to travel back in time to observe the last common ancestor of these two species, this ancestor would NOT have the feature they share. We call this phenomenon (similar yet independently evolved features) convergence, because the two species arrived at the same feature from different evolutionary starting points. There are several explanations for the phenomenon of convergence. One possibility is that the species converged on a particular trait or suite of traits because they both encountered a similar environment and evolved similar adaptations to that environment. Alternatively, the two species could share the same feature for purely non-adaptive reasons. Convergence could occur simply by chance; for instance, if two species have similar physical limits on the types of features their development can actually produce. It is also possible that one species evolved the feature as an adaptation to a particular environment while the other species only possesses the feature because it inherited it from an ancestral species; while the feature may have been an adaptation to a particular environment in this ancestral species, if the modern species no longer encounters the same environment, then the feature is no longer considered an adaptation in the modern species (it’s more like biological baggage that the species is stuck with). At this point, you may be thinking that this is all a nice intellectual exercise, but that there is no feasible way to differentiate between homology and convergence, or between the different causes of convergence in any concrete, scientific way. After all, in order to test hypotheses of homology, convergence and adaptation, we would need to be able to look back thorough time and see when and where features first evolved! Until the later half of the last century (i.e., the 1960s and ‘70s), this was the case: evolutionary ecologists really had no method or theoretical framework for testing these sorts of hypotheses. Around this time, however, the work of a German entymologist named Willi Hennig gained recognition and became a new and powerful tool for testing evolutionary hypotheses. Hennig introduced a method for analyzing the features of species (called characters) to produce an evolutionary diagram called a cladogram. The components of a cladogram and what they represent biologically are described below: In this cladogram, the tips of the lines (denoted by the !, %, #, @ and *) represent the organisms that have been included in the analysis; the cladogram depicts the evolutionary relationships of these organisms. The lines on this cladogram represent evolutionary lineages (usually species) through time, so as you travel from the tips of the lines down to the point where the lines all join (D), you are traveling back through evolutionary time. The points where the separate lines connect are called nodes. Nodes represent the most recent common ancestor or (MRCA) of evolutionary lineages. In this cladogram, the node “D” is the MRCA of all of the species in the analysis. “C” is the MRCA of species !, % and #, while “A” is the MRCA of % and # and “B” is the most recent common ancestor of @ and *. As you can see on the cladogram, nodes are really the points in time where one ancestral species splits into two daughter species. An important point to remember when learning to read cladograms is that they are all about relative ancestry. What this means is that the real information about the relationships between species at the tips of a cladogram is found in the nodes: namely, how many nodes (or lineage splitting events) separate one tip species from another. Because cladograms represent relative relatedness of species, they can’t be fixed in space – in other words, the diagram shown above can be flipped 180 degrees around any of the nodes without changing the relative relatedness (the MRCAs) of any of the species at the tips. It’s key to remember that the order and distance between the species along the tips of the diagram do NOT contain any information about the actual relationships of the species. As an example, in the cladogram above, # and @ appear physically closer to each other along the top of the diagram than # and !, but # and @ are separated by four nodes, while # and ! are only separated by two nodes. This means that # and ! are actually much more closely related (they’re separated by fewer speciation events) than # and @. One final note on cladograms – it is also important to realize that these diagrams really represent a hypothesis of evolutionary relationships. The relationships portrayed by a cladogram can change when additional data or more sophisticated analytical techniques are utilized. For this reason, it is always a good idea to regard cladograms as hypotheses in the process of continual improvement and refinement, and not as static, unchanging fact (this is true of all the products of scientific research). How can cladograms help us test for convergence? Sometimes it can be very hard to decide whether two species that share many features are similar due to common ancestry (homology) or convergence. Because cladograms are created based on the analysis of many different characters (most commonly DNA sequence data in modern analyses), the evolutionary “signal” present in the majority of the characters tends to overwhelm any false indication of evolutionary relationship that might be present in a few convergent characters. This means that once a cladogram has been obtained, the evolution of a particular feature or character of interest can be re-examined in the context of the cladogram. This sort of analysis of character evolution is usually done by “mapping” the presence or absence of the character of interest back onto the species at the tips of the cladogram, and then, based on the relationships of the species on the cladogram, calculating how many times the character would have to either evolve (be gained) or be lost in order to explain its distribution in the tip species. The basic principle used in this process is called parsimony. Simply stated, “parsimony” is the expectation that the simplest answer to a problem is probably the best answer. In our case, this means that if the distribution of a trait in two species on the cladogram can either be explained by two independent gains of the trait or one gain and seven subsequent losses of the trait, the first explanation (which requires only two events as opposed to eight) is preferred. We will explore this idea a bit further using plants that share the feature of stem succulence. The cladogram above represents our most current understanding of the relationships between a large group of flowering plants known as the core eudicots. A group of species (or, in this case, orders of plants) such as this that all descend from a single common ancestor (the node at the base of this cladogram) is called a clade. All of the orders of plants at the tips of this cladogram are themselves clades composed of many species (orders are shown at the tips here, because we would need hundreds of pages to display all the species at the tips!). Can you think of how we might use this cladogram of the core eudicots to determine whether stem succulence in the cacti and the euphorbs is due to homology or convergence? The cacti are part of the Caryophyllales, and the euphorbs are found in the Malpighiales. In order for stem succulence in the cacti and euphorbs to be a homology, the most recent common ancestor of the Caryophyllales and the Malpighiales would have to have possessed this trait. Find the most recent common ancestor of the Caryophyllales and Malpighiales. Now look at the other orders (which don’t contain stem succulents) that also descended from this ancestor. Is it more parsimonious to assume convergence (independent gains of stem succulence in cacti and euphorbs) or homology (one gain and multiple losses of stem succulence) for this trait?