mec12568-sup-0002-FileS1
advertisement

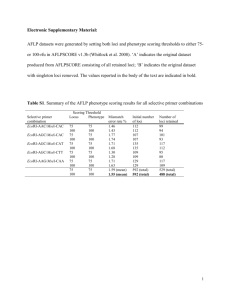
1 File S1: Additional information of methods and parameters used for statistical analyses. 2 3 SUPPLEMENTARY INFORMATION 4 Detection and characterization of outlier SNPs 5 The first analysis approach followed a method implemented in the software Arlequin 3.5 6 (Excoffier & Lischer 2010), based on coalescent simulations to obtain a null distribution of FST 7 or FCT across loci, depending on the assumed demographic model (i.e. finite island or 8 hierarchical island models, respectively; Excoffier et al. 2009), as a function of 9 heterozygosity. Loci showing higher or lower differentiation with respect to the simulated 10 confidence intervals are identified as candidates for divergent or balancing selection 11 (Beaumont & Nichols 1996). Excoffier and colleagues (2009) showed that the hierarchical 12 island model reduces the excess of false positives that arises when samples belonging to a 13 structured population are analyzed under a finite island model. Taking into account the 14 genetic differentiation between Atlantic and Mediterranean populations demonstrated in 15 previous studies, large-scale outlier detection analysis was carried out under the assumption 16 of a hierarchical island model, grouping samples according to the basin of origin. The finite 17 island model was used to analyze the subset of samples within each basin, with 50,000 18 simulations were run and 100 demes per group were assumed, simulating 10 groups under 19 the hierarchical model assumption. 20 The second approach, based on the Bayesian method and implemented in the software 21 Bayescan 2.0 (Foll & Gaggiotti 2008), tests for departure from neutrality by evaluating the 22 weight of the locus-specific contribution with respect to the population-specific effect. The 23 posterior probability for a locus being under selection is calculated according to two 1 24 alternative models, neutrality and under natural selection. This approach appeared to be 25 robust under complex demographic scenarios (Foll & Gaggiotti 2008), and was found to have 26 lower type I (false positive) and type II (false negatives) error rates for divergent selection 27 compared to other outlier detection methods (Narum & Hess 2011). All analyses were based 28 on 20 pilot runs, each consisting of 5,000 iterations, followed by 100,000 iterations with a 29 burn-in of 50,000 iterations. The prior odds for the neutral model was set to 10, as 30 suggested for the identification of candidate loci with a few hundred markers. Posterior 31 Odds (PO), indicating the increased likelihood of the model including selection compared to 32 the neutral model, were interpreted according to the Jeffreys' scale of evidence for Bayes 33 Factors (BF) (Jeffreys 1961). Following this method, a log10PO between 1 and 1.5 denotes 34 strong evidence for selection, between 1.5 and 2 it can be considered as very strong 35 evidence, while values higher than 2 indicate a decisive signal. 36 Correlation analysis between genetic and environmental variation 37 The method first estimates a null model based on neutral markers describing how allele 38 frequencies co-vary across populations, and subsequently it tests whether the correlation 39 observed between allele frequencies at specific markers of interest and an environmental 40 variable is higher than expected under a null model. In this way, the underlying population 41 structure is taken into account, and the probability of obtaining false positive results due to 42 shared population history or gene flow is limited. The software provides the Bayes Factor 43 (BF) as a measure in support to the model including a significant correlation. Putatively 44 neutral, unlinked SNPs were used to calculate the covariance matrix, and then for each 45 candidate SNP the BF was calculated in relation to selected environmental parameters, 46 following the multiple spatial scale approach adopted in outlier analysis. Independent runs 2 47 were carried out to ensure that results were not sensitive to stochastic errors. Results were 48 evaluated comparing the distribution of BF values across putatively neutral and outlier loci 49 and according to the Jeffreys' scale of evidence for BF (Jeffreys 1961). 50 Annual mean values of seawater surface temperature (SST, °C) and salinity (S, psu) were 51 retrieved 52 (www.nodc.noaa.gov/OC5/SELECT/dbsearch.html), referring to the geographic coordinates 53 as close as possible to actual sampling locations for which data were available (Table S1). 54 NODC statistics on SST and S parameters are based on long-term observations (50 years, 55 from 1955 to 2006), and records vary across a quarter-degree latitude-longitude grid 56 (Antonov et al. 2010; Locarnini et al. 2010). To assess the robustness of correlation statistics 57 the annual mean values of temperature and salinity at – 100 m depth were also tested (data 58 not shown). 59 Population genetic structure: neutral vs outlier divergence 60 Structure uses a Bayesian algorithm to infer the number of distinct K clusters of individuals, 61 based on their multilocus genotypes, assuming HWE and Linkage Equilibrium. A posterior 62 probability for each inferred K is calculated, allowing the estimate of the most likely number 63 of clusters. The algorithm was run assuming an admixture model and correlated allele 64 frequencies among populations, as well as providing sampling information as a prior, in 65 order to improve accuracy in detecting population structure (Hubisz et al. 2009). For each 66 analysis 6 iterations were used per K value, a burn-in period length of 100,000, and 500,000 67 MCMC repetitions. To identify the most likely number of clusters accounting for the 68 observed genetic structure the optimal K was selected according to two criteria: the 69 evaluation of the log probability of the data and the Evanno method (Evanno et al. 2005). A from the National Oceanographic 3 Data Center (NODC) database 70 final long run was performed (10 iterations, 500,000 burn-in, 750,000 MCMC repetitions) 71 based on the most probable values of K. Results from multiple runs at the optimal K were 72 combined using the CLUMPP software v.1.1.2 (Jakobsson & Rosenberg 2007), and the 73 graphical output was obtained by Distruct (Rosenberg 2004). 74 To corroborate the genetic structure inferred from Bayesian clustering, the DAPC approach, 75 a multivariate method that does not rely on specific population genetic models, was also 76 used. According to this method, genetic data are first transformed using Principal 77 Component Analysis (PCA) into components explaining most of the genetic variation. These 78 components are then used to perform a linear Discriminant Analysis (DA), which provides 79 variables describing genetic groups, minimizing the genetic variance within populations, 80 while maximizing among-population variation. 81 82 References 83 Antonov JI, Seidov D, Boyer TP, et al. (2010) World Ocean Atlas 2009, Volume 2: Salinity. U.S. 84 Government Printing Office, Washington, D.C. 85 Beaumont MA, Nichols RA (1996) Evaluating Loci for Use in the Genetic Analysis of 86 Population Structure. Proceedings of the Royal Society of London. Series B: Biological 87 Sciences, 263, 1619-1626. 88 89 Excoffier L, Hofer T, Foll M (2009) Detecting loci under selection in a hierarchically structured population. Heredity, 103, 285-298. 90 Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform 91 population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10, 92 564-567. 4 93 94 Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology, 14, 2611-2620. 95 Foll M, Gaggiotti O (2008) A Genome-Scan Method to Identify Selected Loci Appropriate for 96 Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics, 180, 977- 97 993. 98 Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure 99 with the assistance of sample group information. Molecular Ecology Resources, 9, 1322- 100 1332. 101 Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program 102 for dealing with label switching and multimodality in analysis of population structure. 103 Bioinformatics, 23, 1801-1806. 104 Jeffreys H (1961) Theory of probability Clarendon Press, Oxford. 105 Locarnini RA, Mishonov AV, Antonov JI, et al. (2010) World Ocean Atlas 2009, Volume 1: 106 107 108 109 110 Temperature. U.S. Government Printing Office, Washington. Narum SR, Hess JE (2011) Comparison of FST outlier tests for SNP loci under selection. Molecular Ecology Resources, 11, 184-194. Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Molecular Ecology Notes, 4, 137-138. 5