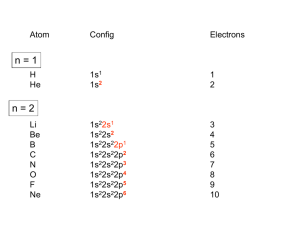
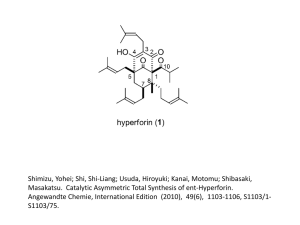
Full text - FNWI (Science) Education Service Centre
advertisement
 Education Service Centre")
Synthesis of C2-symmetric di-N-heterocyclic carbene bidentate ligands and their applications in asymmetric catalysis Master literature thesis by Lukas Jongkind 6116523 Date: 07-10-2013 Daily Supervisor: S.N. Sluijter MSc. Supervisor: Prof. dr. C.J. Elsevier Second Examiner: Dr. J.I. van der Vlugt Molecular Inorganic Chemistry Van ’t Hoff Institute for Molecular Sciences Universiteit van Amsterdam Abbreviations BINAP BINAM COD dba DIBAL-H DMAP DME dr DPE-PHOS ee ndb NHC THF 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl 2,2′-Bis(diamino)-1,1′-binaphthyl cyclooctadiene dibenzylideneacetone diisobutylaluminium hydride 4-Dimethylaminopyridine dimethoxyethane diastereomeric ratio (Oxydi-2,1-phenylene)-bis(diphenylphosphine) enantiomeric excess norbornadiene N-heterocyclic carbene tetrahydrofuran 2 Table of contents 1. Abstract......................................................................................................................................3 2. Introduction................................................................................................................................5 2.1 Asymmetric catalysis.....................................................................................................5 2.2 N-heterocyclic carbenes................................................................................................7 2.3 Asymmetric catalysis using N-heterocyclic carbene ligands.........................................10 3. Synthesis of N-heterocyclic carbene ligands...............................................................................12 3.1 Synthesis of N-heterocyclic carbenes ligands from azolium salts.................................12 3.2 Synthesis of regular N-heterocyclic carbene ligands....................................................14 3.3 Synthesis of chiral N-heterocyclic carbene ligands.......................................................15 3.4 Synthesis of atropisomeric di-N-heterocyclic carbene ligands......................................16 3.5 Synthesis of C2-symmetric Veige-type di-N-heterocyclic carbene ligands.....................19 3.6 Synthesis of other chiral di-N-heterocyclic carbene ligands.........................................21 4. Asymmetric hydrogenation and hydrosilylation reactions using chiral di-N-heterocyclic………..25 carbene catalysts 4.1 Asymmetric hydrogenation of alkenes using Burgess' ligand.......................................25 4.2 Asymmetric hydrogenation of alkenes using C2-symmetric di-N-heterocyclic ..............26 carbene ligands 4.3 Asymmetric hydrosilylation.........................................................................................28 5. Asymmetric addition reactions using chiral di-N-heterocyclic carbene ligand catalysts..............32 5.1 Asymmetric conjugate addition...................................................................................32 5.2 Asymmetric addition reactions to carbonyls and imines..............................................36 5.3 Other enantioselective reactions catalysed by di-N-heterocyclic carbene complex......42 5.4 Selective transformations using di-N-heterocyclic carbene ligands.............................46 6. Conclusion.................................................................................................................................50 7. References................................................................................................................................53 3 1. Abstract N-heterocyclic carbene ligands are relatively novel ligands in organometallic chemistry. Their interesting electronic properties have led to a lot of research into the application of NHC ligands in homogeneous catalysis. Although several effective NHC ligand catalyst are known, a general design motive for chiral NHC ligands has not been identified. Searching for a general ligand design with good definition of chiral space, C2-symmetric di-NHC bidentate ligands have been proposed as one of these possible design motives. Herein, the synthesis and catalytic applications of this class of ligands are evaluated/discussed. Reports of the synthesis of C2-symmetric di-NHC bidentate ligands showed that two different design motives gave promising results. Yields obtained were good and these ligands were easy to modify. To evaluate whether these ligand designs can be a general ligand design for chiral NHC ligands employed in efficient asymmetric catalysis, the catalytic results were reported. These showed that the BINAP-inspired di-NHC complexes reported by Shi et al. gave very good results for numerous enantioselectively catalysed reactions The Shi ligands are easy to synthesise and modify and good catalytic result were achieved with Shi ligand complexes. These ligands are therefore a general and widely applicable ligand design motive for chiral NHC ligands. 4 2. Introduction Asymmetric catalysis is a rapidly expanding area of research within inorganic and organometallic chemistry. Many compounds in nature are chiral. Therefore the need to efficiently induce this chirality onto prochiral substrates, in order to be able to synthesize natural products, derivatives of natural products or drugs, is dire. For most industrial catalytic processes heterogeneous catalysis is used, but because of the complex nature of asymmetric catalysis most catalytic processes within asymmetric syntheses are homogeneously catalysed.1 Research up till now has mainly focussed on chiral phosphine ligands to perform catalytic asymmetric reactions. This is mostly because phosphines bind strongly to metals, and the phosphine ligands are easily modified in terms of electronic and steric properties.2 There is one class of ligands that is frequently compared to phosphines because of their strong σdonating properties and their poor π-acceptor properties, the N-heterocyclic carbene (NHC). Nheterocyclic carbenes are being explored as replacement for phosphines, and are employed in numerous homogeneously catalysed processes, such as Heck,3 Suzuki and Kumada coupling reactions,4 hydrosilylation5 and, probably best known, olefin metathesis (Grubbs’ catalyst).6,7 Asymmetric NHC ligands, although already reported by Lappert et al. in 1983, are currently a relatively unexplored subject compared to the vast amount of chiral phosphine ligands known in literature.8 This literature thesis will focus the design motives available for chiral NHC ligands and the applications of these ligands in asymmetric catalysis. 2.1 Asymmetric catalysis Asymmetric catalysis has been a very import area of research within the area of inorganic chemistry for a long time. Various efficient procedures using asymmetric catalysts are nowadays standard reactions within synthesis. Key examples are Sharpless’ epoxidation procedure and Noyori’s asymmetric hydrogenation catalyst (figure 1).9,10 Figure 1: Sharpless’ epoxidation catalyst and Noyori’s asymmetric hydrogenation catalyst 5 Key to efficient asymmetric synthesis is stabilizing transition states leading to one enantiomer, whilst at the same time disfavouring the formation of the other enantiomer through destabilizing the transitions state leading to that enantiomer. This can be effectively achieved by controlling the coordination of a substrate to the metal centre by “blocking” certain coordination modes, effectively ensuring that the substrate will only coordinate in one fashion to the metal centre (figure 2). Figure 2: Two possible coordination modes for a 1,3-keto ester to bind to the ruthenium-BINAP complex, steric interactions between R1 and the phenyl substituents on the phosphorous groups pointing forwards being the interaction leading to a favourable and unfavourable coordination mode Being C2-symmetric, Noyori’s asymmetric hydrogenation catalyst has the added advantage that the two favourable modes of coordination possible for substrates are equivalent. Because of this, the number of diastereomeric intermediates and transition states is decreased dramatically.11 C2-symetric ligands are therefore very efficient ligands, and a strong basis for design of chiral ligands. As an example, the C2-symmetric BINAP (2,2'-bis(diphenylphosphino)-1,1'-binaphthyl) systems are currently the only chiral ligands produced on an industrial scale, and BINAP complexes are used in a number of large scale processes.12 6 C2-symmetric di-phosphino complexes have shown remarkable success in asymmetric catalysis. Because of the similarities between phosphines and NHC’s, research into these C2-symmetric chiral motives may lead to finding a general and widely applicable ligand design for chiral NHC ligands. 2.2 N-Heterocyclic Carbenes Organometallic N-heterocyclic carbene complexes were first reported by Öfele and Wanzlick et al. in 1968.13 Öfele and Wanzlick reported complexes of imidazole-2-ylidenes, the NHC motive that is used most often in organometallic chemistry. In 1991 Arduengo et al. managed to isolate a stable version of these reactive carbenes.14 This was achieved by strongly increasing the steric bulk on the nitrogen atoms in the imidazole ring, which prevented the dimerization reaction, which normally occur very rapidly in carbenes (figure 3). Figure 3: Dimerization reaction of free carbenes N-heterocylic carbenes are often compared to (tertiary) phosphines because both are strong σdonor and exhibit poor π-acceptor properties. NHC’s are however even stronger σ-donors than tertiary phosphines, which accounts for the fact that the metal-carbene bond is stronger and shorter than the bonds encountered in metal-phosphine complexes.15 This leads to NHC’s having a higher thermal stability than phosphine complexes. NHC’s also exhibit better stability towards oxygen and moisture, as especially trialkyl phosphines tend to be sensitive to oxidation. The electronic properties of NHC ligands are mostly controlled by the location of the nitrogen atoms in the ring. The nitrogen atoms are responsible for stabilizing the carbene through overlap between the lone pairs on the nitrogen atoms and the free orbital of the carbene (figure 4). Figure 4: Orbital overlap and inductive effects in NHC’s 7 Arduengo type NHC’s, imidazole-2-ylidenes, are the most well-known type of NHC’s. There are however several other types of NHC’s, which have slightly different electronic properties (figure 5).16 Figure 5: Electron donating properties of selected NHC’; phosphines are weaker donors than the least electron donating NHC on this scale The nitrogen atoms are, besides being stabilizing through π-overlap, also inductively mildly electron withdrawing, and therefore σ-donation properties increase as the nitrogen atoms are located further away from the carbene. The NHC-ligands that have no nitrogen atom adjacent to the carbene are the strongest σ-donating NHC ligands, and are known as remote NHC’s. Imidazolylidenes are the NHC-ligands mostly encountered in literature. Other types of NHC may however show interesting electronic properties or more facile synthesis routes.17 Other types of NHC's are becoming of greater interest to organometallic chemists, as they prove to be more than academic curiosities. The improved σ-donating properties of NHC’s as compared to phosphines is perhaps best illustrated by one of the most well-known catalyst using a NHC ligand: Grubss’ second generation catalyst for olefin metathesis. Grubbs’ first generation catalyst employs two tricyclohexylphosphine spectator ligands.18 In Grubbs’ second generation catalyst one of these phosphine ligands is replaced by a NHC spectator ligand (figure 6).19 Figure 6: Grubbs’ first and second generation catalyst for olefin metathesis 8 The increase in electron density on the metal caused by the NHC ligand will labilize the metalphosphine bond, facilitating dissociation of the phosphine ligand, which is needed for catalysis. The effect on the catalytic rate of the reaction is significant as the catalytic rate is enhanced by a factor of 102-103.7,20 Considering sterics, NHC’s differ significantly from phosphines.21 It was found that NHC’s are more sterically demanding than most PR3 ligands, and that even small NHC’s, with methyl substituents on the nitrogen atoms are more demanding than P(OMe)3 ligands.21 This may be accredited to the fact that NHC’s are rigid ring structures in which the nitrogen substituents are pointing towards the metal centre and the steric bulk surrounding the metal centre. This is in stark contrast to phosphine ligands, in which the phosphorous atom is surrounded by four groups, three being the R-groups, and the other being its coordinating lone pair. These four groups causes phosphine ligands to have a tetrahedral shape, and as the lone pair of the phosphorous atom coordinates to the metal centre, the R-groups are irrefutably pointing away from the metal, making it more difficult to cause large steric effects, unless one uses very bulky ligands (figure 7). Figure 7: Structure of phosphines and NHC’s showing that NHC’s cause more steric interaction near the metal centre NHC’s tend to form stable complexes with most of the transition metals, with late transition metal complexes of especially ruthenium, rhodium, iridium and palladium being the most abundant in literature.22,23 NHC’s have been reported to form mainly octahedral complexes with d6-metals and square planar complexes with d8-metals, in which the NHC ligand is preferably coordinated trans to a π-acceptor ligand, as the trans effect of the strongly σ-donating NHC ligand is large. NHC complexes may be generated using various methods (see chapter 3.1) starting mostly from metals complexated to weakly coordination ligands such as cyclooctadiene or halide complexes. Di-NHC ligand complexes tend to form cis-coordinated complexes, with most di-NHC complexes preferring bite angles between 79° and 94°.24 Some trans-coordinating di-NHC complexes are known to literature, but because of the flexibility of the linker between the NHC moieties these ligands do not perform well in catalysis.25 9 2.3 Asymmetric catalysis using N-heterocyclic carbene ligands Research into chiral NHC ligands is a relatively unexplored area within organometallic chemistry, despite the first chiral NHC-complex already being reported in 1983 by Lappert et al. Until 2001 only two efficient chiral catalyst containing a NHC ligand were reported, Burgess’ catalyst for asymmetric hydrogenation and Grubbs’ asymmetric olefin metathesis catalyst (figure 8).26,27 Figure 8: Burgess’ hydrogenation catalyst and Grubbs’ catalyst for asymmetric olefin metathesis The chirality in the first NHC-ligands catalysts reported in literature is based on chiral groups attached to the nitrogen atoms of the NHC or to the backbone of the NHC. These ligands are a logical starting point when searching for effective asymmetric catalyst containing NHC-ligands; the simple monodentate ligands are well studied in terms of binding and synthesis, and are known to form stable complexes with most metals. Attaching groups to the nitrogen atoms or the backbone of the NHC does not influence these characteristics in a major way, which makes NHC ligands with chirality on the nitrogen atoms or in the backbone a convenient type of ligand to explore. Reports in literature however indicate that, although convenient research subjects, these ligands are not very suitable to be employed in asymmetric catalysis. When introducing chirality in the backbone, it was found that the chirality is too remotely located with respect to the metal centre where the actual catalysis happens.28 Chirality from the backbone is therefore not efficiently transferred to the substrate, which renders introduction of chiral motives in the backbone of a NHC useless for efficient asymmetric catalysis.28 For NHC ligands with chiral nitrogen substituents a widely applicable catalyst with good results in various asymmetric catalyses has not been reported. This may be accredited to the dynamic nature of these monodentate NHC’s (figure 9). In metal complexes containing these ligands, rotations 10 around the metal-carbene bond and the nitrogen-substituent bonds will cause the chiral space around the molecule to be ill-defined. The lack of a well-defined chiral space makes asymmetric catalysis using ligands with chiral substituents on the nitrogen atoms difficult.28 Figure 9: Structural remarks regarding the location of the chiral groups One way to counteract the dynamic nature of NHC-ligands is to use very bulky substituents that hinder rotation around the nitrogen-substituent bonds. This will however mean that very complex and bulky substituents have to be introduced in a stereoregular way to ensure the successful synthesis of a chiral catalyst.29 This will complicate the system to such an extent that, although useful for some processes, the introduction of chirality on the nitrogen atoms or in the backbone of the NHC will not lead to a simple, yet effective, new design for chiral NHC-ligands in general. One possibility to design a simple but effective chiral NHC ligand is to use bidentate ligands. Bidentate ligands that combine a NHC moiety with another coordinating atom, such as an oxygen or nitrogen atom, are an option for designing a chiral bidentate NHC ligand. Oxygen or nitrogen atoms coordinated to a metal centre are however known to be hemi-labile. These hetero atoms will occasionally dissociate from the metal atom, meaning that the well-defined structure of the complex is lost every so often. The solution may be to use two NHC moieties coordinated to the same metal. These so-called diNHC bidentate ligands are known to form very stable complexes with various metals. Di-NHC bidentate ligands do not show problems with dissociation, and therefore the complex will remain in the same geometric state in all cases. This is important for enantioselective catalysts, as generally only one conformation will participate in effective catalysis. Another advantage of using di-NHC bidentate ligands is the fact that these ligands are entirely C2-symmetric, which makes synthesis of such complexes more facile than for NHC-hetero atom bidentate ligands. Furthermore, C2-symmetric bidentate phosphorous ligands are widespread in literature. A prime example is the before mentioned BINAP-system that was employed by Noyori for his asymmetric hydrogenation catalyst. Because of the similarities between phosphines and NHC’s in terms of electronic properties and the large success of C2-symmetric complexes in asymmetric catalysis this thesis will focus on finding a strong, general ligand design motive for chiral C2-symmetric di-NHC bidentate ligands. 11 3. Synthesis of N-Heterocyclic carbene ligands When it comes to designing an effective asymmetric catalyst, one of the aspects that has to be considered is the synthesis of the complex. Several factors play a role in deciding whether ligands and complexes can be synthesized efficiently. An important factor for the synthesis of a possible catalyst is to design a synthetic route that can be modified easily. When screening catalysts, complexation to a metal atom may go very efficiently, but catalysis may not give good results. In such a situation the catalytic results may be enhanced by increasing the steric bulk of the complex, because that will generally enhance the definition of the chiral space. If one wants to introduce a general and widely applicable ligand motive, changing the sterics should be facile. If it is not easy to change the sterics of a complex without drastically changing the synthetic route towards the complex, finding broad applications for the ligand will be difficult. Another factor that has to be considered is the availability of the starting compounds needed for the synthesis of the desired complex. Especially when synthesizing chiral complexes, one has to consider the fact that most chiral substances are not produced on large scale and/or may be expensive. This may be resolved by using a chiral element that is produced on a larger scale or by choosing to derive the chiral element from a natural compound. Natural compounds are often available in one enantiomer which can be found in nature. As an example, some N-heterocyclic carbene ligands can be derived from amino acids, but one has to consider the fact that the natural amino acids are all Lamino acids. The L-amino acids are therefore widely available, but the D-amino acids are only produced on small scale, and can therefore be expensive. 3.1 Synthesis of N-heterocyclic carbenes from azolium salts Most N-heterocyclic carbenes are prepared via deprotonation of imidazolium salts (scheme 1).30 Most often the azolium salts are deprotonated using strong bases at low temperatures. Depending on the conditions and robustness of the ligand, bases ranging from extremely strong, such as butyl lithium, to less strong bases, such as sodium ethoxide, can be used. The metal is usually added as a complex with weakly coordinating anions, which are easily replaced for the strongly coordinating NHC’s.30 Another method used frequently is the use of an internal base for deprotonation of the azolium salt. In that case one of the weakly coordinating ligands will act as a base and deprotonate the azolium salt. The ligands used have to be basic as a certain base strength is required. One of the most frequently used internal bases is the acetate anion 12 Scheme 1: General procedure for generation of NHC complexes Besides deprotonation there are two more procedures to generate carbenes that are reported frequently in literature. These methods are especially useful when synthesizing di-NHC complexes. Bidentate ligands, having two coordinating moieties, often tend not to coordinate in a chelating fashion, but in such a way that both coordinating moieties are coordinated to separate metal atoms. Because of the very stable metal-carbene bond, this may pose a problem when trying to produce a chelated di-NHC complex. The first procedure is transmetallation. This procedure uses silver oxide to remove the proton, which will lead to silver being coordinated to the carbene moiety. The silver can then be replaced by another metal which forms a more stable bond with the NHC (scheme 2).31 The precipitation of the silver salt is a driving force in the transmetallation reaction. The second procedure is a procedure in which the metal is inserted in either an olefin bond or into a dimerized carbene bond. This may be used to generate di-NHC complexes (scheme 2).32 As preparing NHC complexes from azolium salts is a straight forward procedure the more interesting part of the synthesis of NHC complexes is the synthesis of these azolium salts. Scheme 2: Transmetallation and insertion procedure for creating carbenes 13 3.2 Synthesis of regular N-heterocyclic carbene complexes For symmetric imidazolium salts (precursor for imidazolylidenes) the synthesis is facile, requiring a few reaction steps employing basic organic chemistry (scheme 3).30 The synthesis of azolium salts is very general, allowing the introduction of an R-group as long as that R-group can be attached to an amine group. It must be added however that these reactions are increasingly difficult as the Rgroups get bulkier. Scheme 3: General synthesis of disubstituted imidazolium salts For asymmetric synthesis of imidazolium salts general procedures are well known (scheme 3).30 The synthesis of other azolium salts is very comparable to the first two syntheses mentioned. One interesting subclass of N-heterocyclic carbenes does have fundamentally different synthesis route. 1,2,3-Triazolylidenes can be synthesized using a [3+2]-cyclcoaddition “click” reaction between an azide and an alkyne (scheme 4).17 This step is followed by an alkylation step, allowing for the introduction of various R-substituents in the last step of synthesis. Scheme 4: [3+2]-cycloaddition followed by alkylation to yield a trisubstituted 1,2,3-triazolium salt The syntheses mentioned above are all general procedure to obtain azolium salts. With these procedures chiral groups may also be introduced, allowing for chiral azolium salts with chirality in the nitrogen substituents. These may be deprotonated to yield chiral NHC ligands. These methods 14 therefore represent general methods to either generate chiral NHC ligands or to attach NHC moieties to chiral backbones if one is aiming for bidentate ligands. 3.3 Synthesis of chiral N-heterocyclic carbene ligands The first synthesis of chiral N-heterocyclic carbene ligands dates back to 1983 when Lappert et al. reported chiral NHC ligand complex 8 derived from the amino acid (L)-leucine (7, figure 10).8 Although Lappert was the first to develop chiral NHC ligands, there is no report of the use of his ligands in catalysis. Figure 10: Lapperts first chiral NHC complex, derived from (L)-leucine The first report of a chiral NHC-ligand being used in catalysis was made in 2001 when Burgess reported the use of a chiral NHC-heteroatom bidentate system (Burgess’ ligand, 9).26 Burgess’ publication was followed shortly after by Grubbs who reported the usage of a chiral NHC ligand in asymmetric olefin metathesis (10).7,20 Both these ligands were prepared starting with chiral molecules. In the case of Burgess’ ligand the NHC was attached to the chiral molecule. In the case of Grubbs’ catalyst the chiral molecule itself is formed into an NHC (figure 11). Figure 11: Starting materials for the synthesis the ligands used in Burgess’ catalyst (9) and Grubbs’ catalyst (10) 15 As these catalysts show, the first catalysts designed and developed actually used chiral group attached to either the backbone of the NHC or to the nitrogen atoms. In these cases the chiral space is defined well enough. In the case of Grubbs’ catalyst this is ensure by the use of quite bulky ligands which makes synthesis more difficult. These ligands are derived from chiral molecules that are commercially available, which eliminates the need for difficult asymmetric procedures. The Burgess and Grubbs ligands are therefore perfect examples of ligands for efficient asymmetric catalysis: they are derived from compounds that are easily obtained and they can easily be altered; in Burgess’ ligand the R and Ar groups can be modified, and with Grubbs’ ligand the aromatic groups on the nitrogen atom may also be modified. As mentioned before however, chirality introduced in the backbone or on the nitrogen substituents is usually not very effective in asymmetric catalysis. C2symmetic bidentate ligands are usually better at defining chiral space and may therefore give better results in asymmetric catalysis.28 3.4 Synthesis of atropisomeric di-N-heterocyclic carbene ligands The syntheses of various C2-symmetic bidentate NHC ligands reported in literature shall be discussed here. The first synthesis that will be discussed is the synthesis of a BINAM based di-NHC bidentate ligand first reported in 2003 by the group of Shi (scheme 5).33 Scheme 5: Synthesis of Shi’s ligand (azolium salt)33 The synthesis starts with a palladium catalysed coupling of 2-bromo-nitrobenzene to commercially available BINAM (11, (R)-(+)-1,1′-Binaphthyl-2,2′-diamine). The attached nitro group is reduced using 16 hydrogen with palladium on carbon to yield the free amine groups. Triethyl orthoformate is added to allow for the five membered ring to be formed, after which iodomethane is added to yield benzimidazolium salt 12. The benzimidazolium salt is then complexated to rhodium (scheme 6).33 Scheme 6: Complex formation of Shi’s ligand (13) with rhodium The synthesis of Shi’s ligand shows to be very efficient, with the yield going from BINAM to the benzimidazolium salt being 84% (calculated, over four steps).33 It has to be noted however that two catalytic steps are employed in the synthesis of this ligand, which will increase the cost of the synthesis. Complexation to the rhodium is more difficult as the desired product (13) is only formed with 25% yield. The bimetallic complex is formed in 16%, meaning that little of the ligand is successfully complexated to the metal. Both complexes could be separated by column chromatography using silica. Successful synthesis was confirmed through analysis by 1H-NMR, 13CNMR and X-ray. The synthesis of Shi’s ligand complex shows an efficient route towards a chiral bidentate NHC ligand. Synthesis begins from commercially available compounds, which eliminates the need to use asymmetric procedures to introduce chirality later on in the synthesis. The ligand can also be modified easily by changing the methyl group attached to nitrogen for other groups (such as the more bulky benzyl group).34,35 Complexation of the ligand to rhodium is not very efficient, but the yield in this step could be increased by first complexation the ligand to two silver atoms, which can then be reacted in a transmetallation reaction to yield the mono-rhodium complex. This can ensure a more effective complexation process in most cases.31,36 Two variations on the atropisomeric basis of Shi’s ligand were reported. Hindered rotation is in these cases was based on a 6,6’-dimethoxy-1,1’-biphenyl system, reported by Liu et al. and a 6,6’dimethyl-1,1’-biphenyl system, reported by Chen et al.37,38 In the ligand reported by Chen et al. the nitrogen substituents are replace by coordinating pyridine based groups by reacting the substrate with the correct alkylating agent in the last step of the synthesis of Shi’s ligand (scheme 5).38 17 This kind of alkylation step is usually one of the last steps of the ligand syntheses, which makes the modification of the nitrogen substituents facile. Figure 12: Atropisomeric iridium di-NHC complexes reported Liu et al. (14) and Chen et al. (15) An article by Song et al. reports another variation. The ligand consist of a standard binaphtyl system, but the benzimidazolylidenes are replaced by regular imidazolylidenes. The nitrogen substituents are replaced by ethanoyl groups, which also can coordinate to the metal centre, which is in this case nickel (figure 13).39 Figure 13: Atropisomeric nickel di-NHC complex reported by Song et al. (16)39 As shown, atropisomeric multidentate di-NHC complexes have been synthesized starting from commercially available chiral compounds, with the possibility to vary several structural elements, such as the atropisomeric backbone as well as the nitrogen substituents. It has also been shown that these ligands can form complexes with several metals, such as rhodium, iridium and nickel. These types of ligands are therefore very interesting subjects for catalytic studies. 18 3.5 Synthesis of C2-symmetric Veige-type di-N-heterocyclic carbene ligands A structural basis for C2-symmetric ligands that is reported in literature is the borane skeleton. Ligands of this type were reported by the group of Veige.40 The Veige-type ligands can be synthesized in an enantioselective fashion using literature procedures to obtain the chiral motive, after which the actual NHC moieties can be introduced (scheme 7).41 Scheme 7: Synthesis of Veige’s ligand (benzimidazolium salt) Dicarboxylic acid 17 can be prepared through an asymmetric Diels-Alder reaction, providing the chiral motive in high yields. The Dicarboxylic acid is reduced using lithium aluminium hydride yielding alcohol 18. The alcohol is reacted with triflic anhydride, after which the triflated alcohol groups are substituted for 1-methyl-benzimidazol groups yielding benzimidazolium salt 20. Other NHC moieties could be introduced in this step, allowing for easy variation. The overall yield for the synthesis is 79% (calculated, over four steps).42 Scheme 8: Complexation procedure for Veige’s ligand (21) 19 Complexation is achieved by deprotonating benzimidazolium salt 20, which leads to the formation of a double bond between the carbene moieties. Rhodium is added as a RhI species with weakly coordinating norbornadiene ligands, which leads to rhodium being inserted into the double bond creating the desired complex (21, scheme 8).42 The coordination of the ligands to rhodium was achieved with 76% yield over the two steps, and the successful synthesis of the chiral complex was confirmed through analysis by 1H-NMR, 13C-NMR and X-ray.42 Figure 14: Variations on Veige’s ligand complexes 40,43–45 Among the variations on Veige’s ligand Jeletic et al. report ligand (22) in which the NHC groups are also attached to each other via the nitrogen substituents, which causes the molecule to be less flexible (figure 14).43 Among the variations also normal Imidazolylidenes and 1,2,4-triazolylidenes (25) were reported, and of both the benzimidazolylidenes (24) and the Imidazolylidenes (23) several different R-groups were reported.40 The rigidity of the complex can be altered by removing the methylene groups that connect the borane skeleton to the NHC-moieties. Complexes of these ligands with rhodium, platinum, palladium, iridium and ruthenium were reported, showing that these di-NHC ligands are capable of forming stable complexes with a variety of metals.40 The ligands reported by the Veige group have shown to be ligands that are very versatile in terms of electronic and steric properties. This means that, combined with the fact that stable complexes have been produced with a number of metals, these ligands show great potential to be used in asymmetric catalysis. 20 3.6 Synthesis of other chiral di-N-heterocyclic carbene ligands Among the other types of C2-symmetric N-heterocyclic carbene ligands reported in literature is the C2-symmetric motive based on chiral 1,2-trans-substituted cyclohexanes (25). Optically pure 1,2trans-substituted diamines are easily obtained using standard kinetic resolution methods described in literature.46 This method uses racemic 1,2-trans-substituted diamino cyclohexane to which a chiral acid is added. This will cause one of the enantiomers to form a ammonium salt with the chiral acid, while the other enantiomer will stay in the solution. In this manner these chiral molecules are easily obtained in both configurations. Therefore they form a good basis to generate chiral ligands. For the synthesis of the most simple di-NHC ligands derived from optically pure 1,2-transdiaminocyclohexane two procedures have been published in literature (scheme 9).47,48 These synthetic routes allow for several structural elements to be modified easily, such as the nitrogen substituents and the R’-group in the backbone of the NHC. For the first route, the amine groups of 25 are first reacted with an aldehyde to allow formation of imine 26. This imine is used in a base initiated [3+2]-cycloaddition with tosylmethylisocyanide. This will yield an imidazole ring with a substituent on the 5-position (27). The imidazole ring is alkylated to form the imidazolium salt (31). The product was formed with an overall yield 35% starting from enantiopure diamine 25.47 The second route is more complex. Carbon disulphide is added to form dithiocarbamate groups, which are subsequently methylated with methyliodide, forming compound 28. To this compound an aminoester is added to allow for thiohydantoin groups to form (29). The carbonyl moiety is reduced using DIBAL-H, after which the thiocarbonyl moiety is reduced using hydrogenperoxide and acetic acid, yielding the imidazolium salt (31). The overall yield for this reaction was 35% starting from enantiopure diamine 25.48 In both cases the successful synthesis of the chiral complex was confirmed through analysis by 1H-NMR, 13C-NMR and X-ray. 21 Scheme 9: Two routes towards simple 1,2-trans-diaminocyclohexane inspired di-NHC ligands reported in literature47,48 Several variations on ligands that can be synthesized from optically pure 1,2-transdiaminocyclohexane are also reported (scheme 10).49–51 The chiral 1,2-substituted cyclohexane motive can provide several chiral di-NHC ligands, using basic organic chemistry steps. Because of the similarities between these syntheses and the synthesis of Shi’s ligand, similar modifications can be made to these ligands. The chiral cyclohexane platform is therefore a versatile basis for the design of chiral di-NHC ligands, being easy to obtain, synthesize and modify. Complexation to metal centra was reported for all ligands depicted. Both deprotonation as well as transmetallating procedures were used, with complexes of palladium and rhodium being reported. 22 Scheme 10: Syntheses of three chiral di-NHC ligands derived from 1,2-trans-cyclohexane49–51 Although the 1,2-trans-substituted cyclohexane ring is easily obtained, syntheses of the di-NHC complexes are not efficient enough compared to the high yields obtained when synthesizing Shi and Veige type ligands. This makes these ligands slightly less promising for usage as enantioselective catalysts, when purely considering synthetic aspects. Besides the C2-symmetric ligand motives reported in literature the ligand syntheses of several other C2-symmetric di-NHC ligands were also reported (figure 15).52–55 Of many of these ligands no catalysis is known to literature. This may be accredited to the fact that these ligands are far less rigid than other C2-symmetric di-NHC ligands such as Shi’s ligand. 23 Figure 15: Other C2-symmetic di-NHC ligands reported in literature Of all of these ligands the metal complexes were also reported. Although no variations have been reported on these ligands one may expect, as their synthesis consists of some of the basic steps (see 3.1), similar modifications could be made to these ligands, such as changing the R-substituent or changing the NHC moiety. The strong design and many variations that have been published of the three before-mentioned ligand types make these ligands more interesting for asymmetric catalysis. Another class of C2-symmetric di-NHC ligands are reported in literature.56 These ligands are based on an achiral linker and create a chiral environment by using chiral nitrogen substituents (figure 16). Therefore, these ligands are very similar to the monodentate NHC ligand discussed previously. These ligands shall not be discussed, and the focus will be put on the catalysis using di-NHC ligands with a chiral linker. Figure 16: C2-symmetric di-NHC bidentate ligand R*=chiral substituent Many reports of catalysis with C2-symmetric di-NHC ligands have been made in literature. The results of these catalytic reactions shall be discussed in chapters four and five. 24 4. Asymmetric hydrogenation and hydrosilylation reactions using chiral di-Nheterocyclic carbene catalysts Since the first report of catalysis using chiral N-heterocyclic carbene complexes around 2000, the amount of reports of such ligands in literature has increased steadily. Most research was directed towards finding effective and widely applicable monodentate NHC ligands. It was found however that defining chiral space using monodentate NHC ligands was difficult. Effective catalysis could be achieved using very bulky nitrogen substituents, causing the focus in research to be directed towards finding ways to introduce increasingly bulky substituents in NHC rings.28 Monodentate ligands did not provide a general and easily modifiable method to design chiral NHC ligands. A considerable amount of research was therefore directed towards C2-symmetric di-NHC bidentate ligands. These ligands are better at defining chiral space allowing for ligand design to be directed towards optimization of electronic and steric properties for certain substrates. The first catalytic reaction using di-NHC ligands was reported in 2003.33 Since then several different applications of these C2symmetric di-NHC ligands have been reported. A large portion of this research focussed on asymmetric reduction reactions, such as the hydrogenation of alkenes and the hydrosilylation of ketones. 4.1 Asymmetric hydrogenation of alkenes using Burgess' ligand Current asymmetric hydrogenation procedures are dominated by phosphine ligands, the most notable being Noyori’s catalyst based on the BINAP system. The focus in asymmetric hydrogenation ligands has been mostly on bidentate phosphine ligands up to until around the year 2000 when excellent results were achieved with monodentate phosphorous ligands. A large library of phosphorous ligands exists for an extensive range of substrates. Traditionally the substrate scope of phosphorous ligands for olefin hydrogenation was limited to substrates with polar substituents.28,57 The polar substituents functioned as coordinating groups ensuring correct coordination of the chiral substrate. This allowed for these types of substrates to be hydrogenated with very high yields. Recent studies focussed on finding hydrogenation catalysts that do not require such functionalities. One very effective catalyst for asymmetric hydrogenation employing an N-heterocyclic carbene moiety is Burgess’ catalyst (9). A large number of substrates has been successfully hydrogenated using iridium complexes of Burgess’ ligand, including unfunctionalized alkenes.28 Successful hydrogenation of these alkenes shows the strength of Burgess’ ligand in catalysis, as relatively little hydrogenations of unfunctionalized alkenes were reported in literature (table 1).28 25 Table 1: Selected results in asymmetric hydrogenation using Burgess’ catalyst.58,59 Ar=p-methoxyphenyl Substrate Catalyst loading Yield ee/ dr (0,6%) 99% 98% (0,6%) 99% 97% (0,6%) 99% 96% (0,2%) 90% 40:1 Burgess’ ligand gave very good results in the hydrogenation unfunctionalized E-alkenes with ee’s ranging from 84% to 98%. The hydrogenation of Z-alkenes gave ee’s up to 78%, which was a slight decrease compared to the phosphonite-oxazaline ligands.58 Phosphonite-oxazaline ligands reported in literature gave ee’s ranging from 92% to 99% for the same substrates and better results with Zalkenes.57 The Burgess ligand did show very interesting properties in the iridium catalysed hydrogenation of dienes. Figure 17: Diene substrates for Iridium catalysed hydrogenation60 For diene 32 the iridium catalysed hydrogenation using burgess ligand showed 96% yield of the alkane with 87% ee. For dienes 33 hydrogenation to the alkane yielded the product with up to 96% yield and 99% ee. Hydrogenation of dienes 34 gave the best results with yields up to 100% and ee’s up to 99%.60 These results showed the success of Burgess’ ligands in asymmetric catalysis, as reports of asymmetric hydrogenation of dienes are rare in literature.28 26 4.2 Asymmetric hydrogenation of alkenes using C2-symmetric di-N-heterocyclic carbene ligands Asymmetric hydrogenation using C2-symmetric di-N-heterocyclic carbene ligands was reported by Arnanz et al.53 The ligand employed was a C2-symmetric dioxolane based di-NHC ligand. Catalysis was conducted with gold, palladium and rhodium. The substrates used were several substituted diethyl itaconates (scheme 11). Catalysts loading was 0.5% in all cases (table 2). Scheme 11: Asymmetric hydrogenation of itaconates53 Table 2: Selected results of the hydrogenation of itaconates using various metals complexes of the ligand reported by Arnanz53 R-substituent Hydrogen Metal Au TOF (s-1) 2000 ee 15% (S) Phenyl Au 1250 90% (S) Naphtyl Au 150 95% (S) Hydrogen Rh 258 10% (S) Phenyl Rh 16 99% (S) Naphtyl Rh 10 >95% (S) Hydrogen Pd 45 5% (S) Phenyl Pd 17 98% (S) Naphtyl Pd 2 >95% (S) Varying results were achieved using the C2-symmetric di-NHC ligand as ee’s ranged from 5% to 99%. The general trend showed that increasing the steric bulk of the R-substituent increased the ee, but also led to lower turnover frequencies, thus leading to slower catalysis. They reported that for rhodium and palladium the analogous phosphine complexes gave better turnover frequencies, with turnover frequencies being up to 100% higher.53 For gold complexes, however, the NHC ligands gave much higher turnover frequencies compared to the catalytic activity of a gold-DuPhos complex. Ee’s were very similar for both types of ligands for the more substituted olefins, and in the cases where 27 R=H the phosphine complexes gave higher ee’s.53 A last observation reported by Arnanz et al. was that the gold-NHC complex is stable for over three months and was easier to synthesize and manipulate than the gold-DuPhos complex. 4.3 Asymmetric hydrosilylation Asymmetric hydrosilylation is a very effective way to reduce ketones under mild conditions with little danger of over-reduction. Furthermore, the inexpensive nature of hydrosilanes makes hydrosilylation a very good alternative for hydrogenation reactions.28 There are however little reports in literature of chiral phosphine ligands that are employed successfully in asymmetric hydrosilylation of ketones. Various attempts at designing an effective monodentate NHC ligand for asymmetric hydrosilylation led to the development of a NHC ligand with cyclophane wingtips. As these ligands are very bulky, research has been directed towards the employment of chiral bidentate ligands. In 2003 the group of Shi reported the usage of chiral di-NHC ligand 13 that was employed in rhodium catalysed hydrosilylation of ketones with good results (table 3). The substrates were first silylated, after which the formed molecule was hydrolysed yielding the chiral alcohol as the product (scheme 12). Scheme 12: Hydrosilylation of ketones using catalyst 13 28 Table 3: Selected results of rhodium catalysed asymmetric hydrosilylation of ketones using Shi’s ligand (13)33 Substrate Yield ee 87% 98% 82-93% 95-98% 96% 92% 91% 96% 87% 71% 86% 67% 96% 96% Results for aryl alkyl ketones were good with yields ranging from 82% to 98%. The hydrosilylation of dialkyl ketones showed mediocre ee’s when the alkyl chains were long, but the use of an adamantly group next to the ketone gave very good results, proving that there is no chelating effect of the phenyl ring with the complex, but rather a steric effect causing stereo selectivity. The alkyl chains may not provide enough steric hindrance which may be accredited to their dynamic nature. Another application of Shi’s catalyst was reported by Xu et al. in 2007.61 Catalysts 13 and 35 were used in the asymmetric hydrosilylation of β-keto esters, which were hydrolysed afterwards to yield chiral β-hydroxy esters (scheme 13). The catalytic reaction using catalyst 13 and 35 gave good results (table 4).61 29 Figure 18: Asymmetric reduction of β-keto esters Table 4: Results of the asymmetric hydrosilylation of β-keto esters using catalyst 13 and 3561 Substrate Catalyst Yield ee 13 81% 95% 35 78% 80% 13 90% 95% 35 83% 99% 13 91% 98% 35 88% 98% 13 72% 96% 35 65% 90% 13 87-89% 95-97% 35 80-86% 96-97% Overall the more rigid catalyst 13 gave better results regarding yield, whilst regarding ee’s the results are very comparable. Yields did not seem to be dependent on electronics of the aromatic system next to the keto-moiety, as both the p-methyl as well as the p-chloro phenyl groups gave high yields. Steric factors seem to have a profound effect on the yields, as having a methoxy group in the ortho position led to decreased yields. If the aromatic group is a phenyl ring the yields were slightly lower as well, which may imply that an optimum exists regarding steric interactions. In 2009 Liu et al. reported the usage of a catalyst derived from Shi’s catalyst (36) in the hydrosilylation of ketones, and both α- and β-keto esters.37 Catalytic hydrosilylation and hydrolysis yielded the following results (table 5). 30 Table 5: Results of hydrosilylation using catalyst 36 reported by Liu et al.37 Substrate Yield ee 74% 70% 85% 59% 83% 94% 86% 92% 81% 98% 68% 70% 70% 96% 67% 20% Although the yields were lower than with catalyst 13, ee’s were comparable for both catalysts. These results did however show that changing the backbone of the catalyst does not have a very large effect on catalysis. This implies that the catalyst will retain its activity when the atropisomeric backbone or the nitrogen substituents are changed. The substrate scope is also large although not all substrates gave good results. Relatively little research has gone into modifying the ligands used. As several modifications can be made, such as the introduction of different nitrogen substituents or another atropisomeric backbone, further research may lead to extending the substrate scope and the successful hydrosilylation of more substrates. 31 5. Asymmetric addition reactions using chiral di-N-heterocyclic carbine ligand catalysts C2-symmetric di-N-heterocyclic carbene complexes have been reported on several occasions in asymmetric reduction reactions. Other reactions for which relevant catalytic results were reported were asymmetric addition reactions. These addition reactions include conjugate additions, additions to aldehydes and imines, fluorination, dihydroxylation and hydroamination. The last two reactions were not carried out in an enantioselective fashion, but as they are selective reactions, the results will be reported. 5.1 Asymmetric conjugate addition Among the several types of addition reactions for which chiral di-N-heterocyclic carbene ligands are used asymmetric conjugate additions are a relatively well explored class. Conjugate addition reactions have occupied organometallic chemists for a long time, since there are no reports of a generally applicable catalyst for conjugate additions that is capable of converting broad ranges of substrates with good results.28 Research into chiral di-NHC ligands may provide such a catalyst. The first report in literature was made in 2005 by Clavier et al.62 Among the ligands employed by Clavier et al. was a di-NHC complex with a chiral group in the backbone (37, scheme 13). Scheme 13: Cupper catalysed asymmetric conjugate addition using ligand 37 62 Ligand 37 was used in a cupper catalysed conjugate addition of an ethyl group to a cyclic enone. Results were poor however, with two attempts leading to ee’s of 3% and 5% respectively. The article compared several similar ligands in which one or both of the coordinating NHC-moieties were replaced by phosphorous, nitrogen or oxygen coordinating groups. The comparison showed that the di-NHC had the lowest ee’s of all ligands. It also showed that the di-NHC ligand had the best activity of all complexes. As the chirality is somewhat remotely located from the catalytic centre the usage of a stronger chiral motive may lead to an active and selective catalyst. Another factor influencing catalysis in this case is the size of the metallacycle formed. When the di-NHC ligand is used, a seven 32 membered ring is formed upon complexation. When one of the NHC’s is replaced by a phosphorous, oxygen or nitrogen atom, the metallacycle will be six membered. If both coordinating moieties are replaced, a five membered ring will form. These variations in ring size may cause the profound effect on the catalysis. In 2008 the group of Shi reported the use of their ligand in a catalytic asymmetric addition to cyclic enones using a palladium complex of their previously reported ligand.63 The catalyst was used in the addition of several aromatic groups to 2-cyclohexenone and 2-cycloheptenone through the use of boronic acids (scheme 14). Results of the reactions are summarized below (table 6). Scheme 14: Addition of arylboronic acids to cyclic enones Table 6: Results of the asymmetric addition of arylboronic acids to enones63 n= 1 Aryl group 3-Me-C6H4 Yield 97% ee 97% 1 4-Me-C6H4 89% 92% 1 3-MeO-C6H4 90% 97% 1 4-MeO-C6H4 82% 94% 1 2-naphtyl 99% 97% 1 4-C6H5-C6H4 97% 93% 1 3-Cl-C6H4 78% 88% 2 C6H5 88% 91% 2 4-Me-C6H4 90% 91% 2 3-MeO-C6H4 86% 96% 2 2-naphtyl 99% 97% The results showed good to excellent yields and ee’s for most aryl groups. The only exceptions were reported when an electron withdrawing group was used on the aryl group. In the case of m-chloro phenyl the yield and ee were significantly lower than in the other cases. This was the only electron 33 withdrawing group reported.63 The best results were acquired for the most bulky group, the 2naphtyl group. A variation of the trifluoro acetate ligands was used in catalyst 38 by using regular acetate groups. When the authors used the acetate groups it did not lead to significant changes in yields or ee’s in catalysis. The group of Shi also used the same catalyst (38) for asymmetric addition of arylboronic acids to 2,3dihydro-4-pyridones (scheme 15).64 Results of these reactions are summarized below (table 7). Scheme 15: Addition of arylboronic acids to 2,3,dihydro-4-pyridones Table 7: Results of the asymmetric addition of arylboronic acids to 2,3,dihydro-4-pyridones64 Substrate R-group Benzyl Aryl group C6H5 Yield 88% ee >99.5% Benzyl 4-Me-C6H4 85% 96% Benzyl 3-Me-C6H4 80% 95% Benzyl 4-MeO-C6H4 78% >99.5% Benzyl 3-MeO-C6H4 76% 99% Benzyl 2-naphtyl 85% 98% Benzyl 4-C6H5-C6H4 94% 97% Ethyl C6H5 92% 87% Ethyl 2-naphtyl 85% 97% Ethyl 4-C6H5-C6H4 95% 97% t-Butyl C6H5 82% 99% t-Butyl 2-naphtyl 80% 97% t-Butyl 4-C6H5-C6H4 95% >99.5% 34 Good yields an excellent ee’s were achieved using catalyst 38, which shows that the substrate scope of catalyst 38 is wide, as both aliphatic cyclic enones as well as cyclic enones containing other polar groups are arylated in good yields and ee’s. Furthermore, a large number of aromatic groups can be introduced by an unmodified catalyst, which implies the substrate and addition scope may be expanded even further if modifications are made to the catalyst allowing for these modifications. Figure 17: Rhodium catalyst (24b) employed in asymmetric conjugate addition The Veige ligands were also employed in catalytic addition reactions of arylboronic acids.25,42,45 Several variations of Veiges ligand were used for rhodium catalysed conjugate additions.25,45 There is one report of catalyst 24b being employed in the conjugate addition of phenyl boronic acid to 2cyclohexenone (figure 17). Results for this reaction were promising, as after optimization 86% yield and 82% ee was achieved.25 The other rhodium catalysts reported in similar reactions gave bad results.25 Problems were accredited to the fact that according to the authors the balance between finding a complex that lack enough steric interactions to effectively induce chirality and between a complex that is too sterically hindered.45 Further research in the group of Veige was directed towards several variants of a palladium complex of the Veige ligand (39), of which one was used in the addition of arylboronic acids to 2-cyclopentanone and 2-cyclohexanone (scheme 16). The following results were reported (table 8).42 Scheme 16: Addition of arylboronic acids to 2-cyclopentanone and 2-cyclohexanone 35 Table 8: Results of the addition of arylboronic acids to 2-cyclopentanone and 2-cyclohexanone42 n= 2 Aryl group C6H5 Yield >98% ee 46% 2 4-F-C6H4 95% 44% 2 2-naphtyl 95% 40% 2 2-Me-C6H4 62% 33% 2 1-naphtyl 48% 30% 1 C6H5 87% n.a. 1 2-Me-C6H4 36% 50% 1 1-naphtyl 24% 30% The results of the asymmetric addition of arylboronic acids were poor. Yields were good with some combinations of substrates. Ee’s were low, which may be accredited to the dynamic structure of the ligand.25 According to the authors the methylene groups between the borane skeleton and the NHC moieties allowed for too much movement and too little definition of the chiral space. With this type of reaction the sturdiness of ligands such as Shi’s ligand will provide a well-defined and relatively undynamic system which is required for successful induction of chirality.25 The authors did previously report the use of the more rigid catalyst 24b that lacks the dynamics that may be introduced when the methylene linker is used between the borane skeleton and the NHC moiety. This catalyst gave good results in rhodium catalysed conjugate additions, but no reports are made using this ligand in palladium catalysed conjugate addition. 5.2 Asymmetric addition reactions to carbonyls and imines Asymmetric addition reactions to carbonyls and imines are powerful tools to generate chiral alcohols and amines. The vast amounts of possible substrates that may be used in these type of reactions cause a constant need to design and optimize reaction conditions and catalysts. The use of chiral diNHC complexes in these addition reactions has been reported by several groups. Most groups report the addition of aryl boronic acids using cationic palladium complexes. The first report of an asymmetric arylations was reported by the group of Shi in 2009.35 The catalyst used in this report is a di-aqua palladium complex of Shi’s ligand. Several aromatic groups were added to tosyl imines to yield chiral tosyl amines (scheme 17). In first instance the authors decided to vary the R-group and use phenyl groups to add to the imine (table 9). 36 Scheme 17: Addition of aryl boronic acids to tosyl imines using catalyst 40a Table 9: Results of asymmetric addition of phenyl boronic acid to various tosyl imines35 R-group 4-Cl-C6H4 Ar-group C6H5 Yield 99% ee 90% 3-Cl-C6H4 C6H5 97% 82% 2-Cl-C6H4 C6H5 99% 90% 4-Br-C6H4 C6H5 64% 60% 3-Br-C6H4 C6H5 85% 94% 2-Br-C6H4 C6H5 93% 84% 4-F-C6H4 C6H5 99% 94% 2,4-Cl2-C6H3 C6H5 96% 90% 2,3-Cl2-C6H3 C6H5 96% 86% 4-Me-C6H4 C6H5 99% 90% 4-MeO-C6H4 C6H5 99% 88% 2-MeO-C6H4 C6H5 99% 92% 4-NO2-C6H4 C6H5 99% 84% 3-NO2-C6H4 C6H5 99% 81% 2-NO2-C6H4 C6H5 85% 85% 1-naphtalene C6H5 95% 90% 2-furanyl C6H5 99% 80% thiophene-2-yl C6H5 87% 83% Yields and ee’s were high for a large range of substrates, both with electron rich and electron poor aromatic rings. The only R-group giving disappointing results was the p-bromo phenyl group, which gave only 64% yield and 60% ee. All other results were good to excellent. The authors decided to try and expand the scope even further by varying the aryl group (table 10). 37 Table 10: Results of asymmetric addition of various aryl boronic acid to tosyl imines catalysed by 40a35 R-group 4-Cl-C6H4 Ar-group 4-MeO-C6H4 Yield 90% ee 84% 4-Cl-C6H4 3-Me-C6H4 92% 84% C6H5 4-F-C6H4 90% 81% C6H5 4-Ph-C6H4 80% 93% 2-Cl-C6H4 4-CF3-C6H4 99% 86% 2-Cl-C6H4 3-Cl-C6H4 99% 91% 1-naphtyl 2-naphtyl 92% 91% 1-naphtyl 4-Ph-C6H4 95% 90% CH3CH2CH2 C6H5 64% 66% cyclohexyl C6H5 99% 85% cyclohexyl 4-Ph-C6H4 80% 94% cyclohexyl 2-naphtyl 65% 94% The results of varying the aryl group yielded the products in good to excellent yields and ee’s. Changing the R-group of the tosyl imine for alkyl groups caused the yields to decrease slightly, especially in the case of R=n-propyl. The catalyst (40a) showed tolerance for a considerable range of substrates, effectively combining electron poor and electron rich substrates as well as effectively catalysing reactions between electron poor substrates. The catalyst may therefore be applied to from a large range of chiral amines, providing a mild and effective route towards these widely used molecules. A very similar catalyst (40b) was used in the arylation of Boc-protected imines. This reaction was reported by Liu et al.65 The imine was generated in situ through a base catalysed elimination reaction of an αcarbamoyl sulphone moiety, yielding a Boc-protected imine to which the aryl group was added (scheme 18). The authors first reported the addition of phenyl boronic acid to various Boc-protected imines (table 11). Scheme 18: Asymmetric addition of aryl boronic acids to Boc-protected imines 38 Table 11: Results of the addition of phenyl boronic acids to Boc-protected imines using catalyst 40b65 R-group 4-Cl-C6H4 Ar-group C6H5 Yield 75% ee 83% 4-CF3-C6H4 C6H5 83% 87% 4-F-C6H4 C6H5 89% 86% 4-MeO-C6H4 C6H5 52% 86% 3-Cl-C6H4 C6H5 87% 73% 3-Me-C6H4 C6H5 71% 82% 3-MeO-C6H4 C6H5 88% 82% thiophene-2-yl C6H5 84% 84% cyclohexyl C6H5 <5% - The asymmetric addition of phenyl boronic acids to Boc-protected imines gave good results in most cases, regarding yield and ee. The catalysis worked equally well for electron poor R-groups as for electron rich R-groups. The only R-group that gave very low yield of a racemic product was the cyclohexyl group, which is more flexible than the rigid benzene rings used in all the other cases. Alongside the large difference between electronic properties of a cyclohexyl and an aryl group, this may explain why the results of this reaction are so diverse. The positive results acquired prompted the authors to test whether the catalyst would also tolerate the use of different aryl groups (table 12). 39 Table 12: Results of the addition of various aryl boronic acids to Boc-protected imines using catalyst 40b65 R-group Ar-group Yield ee C6H5 4-Cl-C6H4 72% 90% C6H5 4-CF3-C6H4 65% 88% C6H5 4-F-C6H4 87% 86% C6H5 4-MeO-C6H4 77% 76% C6H5 4-Me-C6H4 77% 70% C6H5 3-Cl-C6H4 79% 78% C6H5 3-MeO-C6H4 62% 82% Varying the aryl groups had a negative effect on the results of the catalysis. Yields and ee’s acquired were moderate to good for both electron rich and electron poor aryl groups, showing that catalyst 40b is tolerant to most electronic effects in this reaction. These reactions showed that this catalyst can be employed in the generation of chiral imines with various substituents. Starting from various protected imines, these palladium catalysed asymmetric additions are examples of chiral di-NHC catalyst employed in a general and valuable process. With the principles of asymmetric addition of aromatic groups to Boc-protected imines the authors chose to test the catalyst in the enantioselective addition of cyclic β-ketoesters to Boc-protected imines (scheme 19).66 These type of reactions catalysed by palladium are well reported in literature and successful catalysis with catalyst 40b will prove the true power of this di-NHC ligand (table 13). Scheme 19: Asymmetric addition of β-keto ester to Boc-protected imines 40 Table 13: Results of the addition of β-keto ester to Boc-protected imines66 Ring size Five membered R-group 2-Cl-C6H4 Yield 80% dr 1:5 ee 73% Five membered 3-Cl-C6H4 95% 1:20 91% Five membered 3-CF3-C6H4 90% 1:20 94% Five membered 4-Cl-C6H4 91% 2:1 90% Five membered 4-CF3-C6H4 90% 1:10 95% Five membered 4-F-C6H4 92% 2:1 83% Five membered 4-Br-C6H4 89% 2:1 70% Five membered 4-Me-C6H4 91% 2:1 80% Five membered cyclohexyl 92% One isolated 96% Six membered 3-CF3-C6H4 85% 1:10 88% Six membered 4-CF3-C6H4 79% 1:5 80% Results of this complex reaction were good. In the case in which the imine substituent was a cyclohexyl group, the results were excellent: only one diastereomer was isolated in 92% yield with 96% ee. Other R-groups led to good yields and ee’s and moderate diastereomeric ratios. Five and six membered rings gave comparable results in catalysis. The reaction between electron rich and electron poor substituents gave results that were equally well. The authors also note that different stereochemistry is observed when using the di-NHC complex when compared to using the phosphine ligands, while the backbone used was the same and had the same absolute configuration. This interesting observation was not explained by the authors, which makes it an unresolved curiosity that remains for this reaction. A very similar catalyst to the ones used in addition reactions to imines was also used by the group of Shi for addition reactions to carbonyl species (41).67 Catalyst 41 was employed in an asymmetric allylic umpolung addition reaction (scheme 20). This means that this addition reaction is an electrophilic addition reaction to an aldehyde, whereas normally aldehydes are attacked by a nucleophile in addition reaction. Results are summarized below (table 14). 41 Scheme 20: Asymmetric allylic umpolung addition Table 14: Results of the asymmetric allylic umpolung addition to various aldehydes67 R-group 4-Cl-C6H4 Yield 93% syn:anti 90:10 ee 64% 2-Cl-C6H4 87% >99:1 61% 3-Cl-C6H4 96% 97:3 66% 4-CF3-C6H4 89% 96:4 64% 4-Me-C6H4 81% 96:4 58% 4-MeO-C6H4 74% 90:10 62% 1-neopentyl 96% 97:3 54% heptyl 61% 84:16 61% C6H5(CH2)2 58% 90:10 62% Results showed that electronic properties of the substrates did not have much influence on the catalytic performance. Both electron rich and electron poor aldehydes gave similar yields, syn:anti ratios and ee values. Yields were moderate to high for all substrates. It should be noted that the bulkiest group used (neopentyl) also gave the highest yields. Syn:anti ratios were good with the syn product being the most prevalent in all cases. Enantiomeric excess values were low, as enantiomeric induction is difficult in these types of reactions, as any chirality will be lost when the acetate group is eliminated and an allylic system is generated. This will cause a loss of any chiral information in the allylic substrate. The group of Shi also reported the usage of a palladium complex of Shi’s ligand in the arylation of aromatic aldehydes.68 The products of these reactions can be of great importance to the pharmaceutical industry. Results were however not satisfactory, although yields up to >99% were reported. The ee values reported ranged from 0% to 65%, which was well below the maximum value of 87% reported in literature for similar substrates.69 According to the authors subtle electronic properties play an important role in these types of reactions.68 42 5.3 Other enantioselective reactions catalysed by chiral di-N-heterocyclic carbene complexes Beside the various reports in literature on di-N-heterocyclic carbene complexes being employed in asymmetric hydrogenation, hydrosilylation and conjugate addition reactions, there are also reports of di-NHC complexes being reported in other asymmetric addition reactions. Because these reactions will show the wide applicability of chiral di-NHC ligands, the results of these reactions will be discussed. The palladium di-aqua complex reported in the arylation of imines (40a) was also employed by the group of Shi in the asymmetric arylation of cumulenes (scheme 21).70 Cumulene derived chiral molecules have been found in nature, which makes synthesis of these compounds an interesting target.71 However, no procedure existed at the time of publishing. The results of the first report of asymmetric transformations of cumulene have been summarized below (table 15). Scheme 21: Asymmetric arylation of cumulenes 43 Table 15: Results of the addition of aryl boronic acids to cumulene derivatives70 R-group C6H5 Aryl group 4-Me-C6H4 Yield 95% ee 93% C6H5 4-MeO-C6H4 87% 91% C6H5 4-Et-C6H4 93% 92% C6H5 4-tBu-C6H4 85% 92% C6H5 4-Ph-C6H4 95% 94% C6H5 4-Cl-C6H4 81% 87% C6H5 3-Me-C6H4 88% 92% C6H5 2-naphtyl 91% 92% C6H5 thiophene-3-yl 91% 88% C6H5 thiophene-2-yl 51% 88% C6H5 2-trans-phenylvinyl 67% 50% 4-F-C6H5 4-MeO-C6H4 91% 91% 4-F-C6H5 3-MeO-C6H4 92% 93% 4-Cl-C6H5 C6H5 93% 91% 4-Cl-C6H5 2-naphtyl 92% 93% 4-Br-C6H5 4-MeO-C6H4 69% 92% 4-Br-C6H5 4-MeO-C6H4 89% 86% 3-Br-4-MeO-C6H5 C6H5 91% 85% Results regarding yields and ee’s were moderate to very good for most substrates. A large range of substrates was used, using electron rich and electron poor R-groups and aryl substituents. Steric hindrance within the aryl group had no negative effects on the catalysis, as both the 4-tBu-C6H4 and the 2-naphtyl group gave good results in catalysis.70 The authors reported that the catalyst was successfully employed by them in the transformation of cumulenes into precursors for natural products. The chirality introduced in the catalysed asymmetric addition of aryl boronic acids was successfully retained, showing that the reaction described may be an efficient transformation step in total syntheses.70 In 2009 the group of Shi reported the usage of a palladium complex of Shi’s ligand for a FriedelCrafts reaction of indole with aromatic imines (42, scheme 22).34 The ligands used is a tridentate ligand, as one of benzyl group also coordinates to the metal centre. This bond is broken once the catalytic cycle commences.34 Effective stereocontrol of this reaction is important because both 44 possible enantiomers show biological activity and may therefore be of interest to the pharmaceutical industry.72 If the formation of a certain enantiomer can be controlled through the modification of the electronic parameters of the ligands rather than changing the configuration of the atropisomeric backbone entirely this would be a big step towards efficient syntheses of both of the enantiomers (table 16). Scheme 22: Friedel-Crafts reaction between indole and aromatic imines Table 16: Results of the Friedel-Crafts reaction between indole and aromatic imines34 Aryl group 4-Cl-C6H4 Catalyst 42a Yield 87% ee 54% 4-Cl-C6H4 42b 89% -74% 3-Cl-C6H4 42a 80% 64% 3-Cl-C6H4 42b 82% -66% 2-Cl-C6H4 42a 74% 24% 2-Cl-C6H4 42b 80% 66% 4-F-C6H4 42a 77% 56% 4-F-C6H4 42b 81% -48% 3-F-C6H4 42a 73% 58% 3-F-C6H4 42b 72% -62% 4-NO2-C6H4 42a 71% 48% 4-NO2-C6H4 42b 74% -66% 2,3-Cl2-C6H3 42a 71% 30% 2,3-Cl2-C6H3 42b 78% 60% Although the yields were moderate to good for this reaction, ee’s were low/disappointing. It was however shown that a small modification to the phenyl nitrogen substituents could cause the stereochemistry to reverse, while the chiral backbone was the same in both reactions. This showed that stereochemistry could effectively be controlled by changing the electronic properties of this complex. Before applications can be found however, catalytic yields should be improved. 45 In 2012 the group of Shi reported the use of a palladium di-aqua complex in an asymmetric fluorination reaction (40a).73 Asymmetric fluorinations were discovered in 2000 by Togni et al. and have developed quickly since then.74 Fluorination reactions to create fluorinated tertiary carbon atoms are rare. The group of Shi therefore tried to fluorinate oxindole substrates because these fluorinated substrates may have biological activity (scheme 23).73,74 Such a reaction would also show the wide applicability of Shi’s ligand in catalysis (table 17). Scheme 23: Asymmetric fluorination of oxindoles Table 17: Results of the asymmetric fluorination of indoles73 R-group Me Aryl group C6H5 Yield 96% ee 47% OMe C6H5 97% 44% F C6H5 98% 59% H 2-naphtyl 95% 42% H 3-F-C6H4 92% 48% H 4-Me-C6H4 98% 34% H 4-F-C6H4 95% 22% Me 4-Me-C6H4 90% 45% Me 4-F-C6H4 95% 28% OMe 4-Me-C6H4 98% 49% OMe 4-F-C6H4 94% 38% F 4-Me-C6H4 95% 40% F 4-F-C6H4 95% 20% H 2-Me-C6H4 88% 21% H 3-Me-C6H4 96% 44% H 3,5-Me2-C6H3 96% 35% Yields of the asymmetric fluorination were high, although ee values are low. The results for electron donating and electron withdrawing substituents were comparable, which showed that the catalyst is tolerant towards electronic effects. Steric induction in these indole systems is not facile because 46 differentiation between the two possible faces on which the system can be fluorinated is difficult, since the system is mostly planar. The article proved however that asymmetric fluorination is possible using chiral di-NHC complexes. The usage of another substrate or further modification of the ligand may increase ee’s for these types of reactions. 5.4 Selective transformations using di-N-heterocyclic carbene ligands Besides the numerous reports of asymmetric catalysis using di-NHC ligands there are also several reports of selective transformations using di-NHC ligands that show good diastereoselectivity or regioselectivity. Relevant examples are discussed below. In 2010 the group of Shi reported the use of a palladium di-aqua complex of Shi’s ligand (40b) in the dihydroxylation of alkenes.75 Asymmetric procedures such as Sharpless’ asymmetric dihydroxylation procedure are common procedure within synthesis procedures.76 Recently however there have been reports of difunctionalizations using palladium catalysts,77,78 prompting the authors to try whether palladium di-NHC complexes could be used to form an addition to the already available dihydroxylation procedures (table 19).75 After the reaction acetic anhydride was added which transformed the alcohol groups into acetate groups. Table 19: Results of the palladium catalysed dihydroxylation of alkenes75 Substrate Product Yield syn:anti 79% 8:1 99% 4:1 99% 10:1 64% - 60% 1.5:1 47 The results of the palladium catalysed dihydroxylation of various alkenes were moderate. Yields were moderate to excellent, syn:anti ratios were moderate and no chiral induction was reported. This means that the formed diacetates are still racemic. The authors proved the principle of palladium catalysed dihydroxylation using di-NHC ligand complexes, but the catalyst has to be modified severely before efficient catalysis may be expected. Figure 19: Palladium catalyst used in hydroamination reactions In 2012 the group of Shi reported the use of a palladium complex in a hydroamination reaction with Markovnikov selectivity (45, figure 19).79 Hydroamination reaction of amines with unactivated alkenes are very atom efficient routes towards synthesis of heterocycles. containing nitrogen atoms.79 Although several catalyst exist for hydroamination reactions, the substrate scope of this reaction is very large and further research can be conducted. Several intramolecular hydroamination reactions with unactivated alkenes were performed (scheme 24). The results are summarized below (table 20). Scheme 24: General intramolecular hydroamination of unactivated alkenes The results of the palladium catalysed hydroaminanation were good, with yields all upwards of 85% in the right Markovnikov selectivity. Electron rich and electron poor substituents on the amine group gave comparable results, showing that the catalyst is resilient towards small electronic changes in the substrate. Both five and six membered heterocycles were successfully closed. The results of the hydroamination reaction showed that the di-NHC catalyst is a very widely applicable catalyst that is 48 capable of converting large ranges of substrates. The results of the hydroamination showed no ee which means the products formed were racemates. The catalyst should therefore be modified into an asymmetric hydroamination catalyst before any use in synthesis may be expected. Table 20: Results of the palladium catalysed hydroamination of unactivated olefins79 Substrate R= C6H5 R= 4-Br-C6H4 R= 4-CN-C6H4 R= 4-NO2-C6H4 R= 4-CO2Me-C6H4 R= 4-Me-C6H4 R= 4-OMe-C6H4 R= cyclohexane R= tosyl Yield 99% 93% 95% 94% 92% 90% 85% 92% 90% 85% 90% 85% 90% 88% 90% 90% 92% 95% 49 6. Conclusion Phosphine ligands have long been the ligand of choice for asymmetric catalysis. Since the introduction of N-heterocyclic carbenes an increasing amount of research has been devoted to application of NHC’s in catalysis. Several applications were found for NHC’s owing to their remarkable σ-donating properties and high thermal stabilities. The possibilities of applying NHC ligands in asymmetric catalysis were explored. This led to a variety of chiral monodentate NHC ligands that could be applied in various enantioselective reactions. The design strategy for monoNHC ligands was however not very general, as the focus was mainly on creating a chiral space that was defined well enough to transfer the chirality from the complex to the substrate. This prompted researchers to introduce ever bulkier groups, rather than trying to optimize a ligand for the conversion of a certain substrate. The search for a more general ligand design for chiral NHC ligands led to the introduction of chiral C2-symmetric di-NHC bidentate complexes. The more rigid C2-symmetric bidentate ligands give a better definition of chiral space which eliminates the need for very bulky substituents. The focus could therefore be directed towards modifying the catalysts for certain reactions. The syntheses of the chiral C2-symmetric di-NHC bidentate ligands reported in this thesis show that the routes towards these ligands start with commercially available chiral molecules. These can be transformed into di-NHC ligands using simple organic transformations. The ligands can be modified through the introduction of nitrogen substituents. If the nitrogen substituents are introduced in the last stages of the synthesis of the ligand it makes modification more facile. The ligands can form complexes with numerous metals through various complexation methods and can therefore be employed in several catalytic reactions. In terms of efficient synthesis two C2-symmetric di-NHC bidentate ligands gave very promising results. Shi’s ligand, based on the atropisomeric 1,1’-binaphtyl skeleton, can be synthesised starting from commercially available BINAM with 84% yield over four steps. Many variation are reported for Shi’s ligand. These include variations on the atropisomeric framework and variations regarding the nitrogen substituents. Nitrogen substituents can be introduced in a final alkylation step, allowing for easy modification. Variations of the nitrogen substituents are reported that can coordinate to the metal centre, creating tetradentate ligands allowing for variations in the coordination modes of these complexes. Veige’s ligand, based on a borane skeleton, can be synthesised starting from a fumaric acid in 79% over four steps. The synthesis reported allows for the introduction of the NHC-moieties in one of the 50 last steps of synthesis. The NHC-moieties can therefore easily be modified. Nitrogen substituents can be introduced through alkylation and numerous groups are reported. One interesting variation is reported in which the nitrogen substituents were attached to each other, allowing for a more rigid catalyst. The Shi and Veige ligands are the strongest design motives for C2-symmetric di-NHC bidentate ligands, as they are synthesised in good yields and show great modifiability. Catalysis with complexes these ligands and other C2-symmetric di-NHC bidentate ligands have shown whether these ligands were truly an effective and general design motive for asymmetric catalysis. A large amount of the successful catalysis preformed with C2-symmetric bidentate ligands is reported by the group of Shi. The Shi ligands are used for selective hydrosilylations, conjugate additions, additions to imines, carbonyls and cumulenes, fluorinations, dihydroxylations and hydroaminations. In these reactions the Shi ligands show to be tolerant to a large range of different substrates, both with electron donating groups as well as electron withdrawing groups. The Shi ligand can be employed with relatively little modification in very different reactions, showing that this design motive is a strong design motive for chiral NHC ligands. The Veige type ligands are used for asymmetric hydrogenation and conjugate additions. Results for these ligands are disappointing. Low ee’s are reported for most reactions, the highest ee reported being 82% in an asymmetric conjugate addition. The authors suggest that subtle variations in the steric properties of the complexes causes the bad results, as they report that it was difficult to find a balance between steric hindrance and sufficient definition of chiral space. Rigidity of the ligands also contributes in major way, as in all cases, and especially for these ligands, low rigidity equals low enantioselectivity. Because Veige’s ligands lack good results in asymmetric catalysis, the ligand design motive cannot be considered a general and effective design for chiral NHC ligands. Of all other C2-symmetic di-NHC bidentate ligands no efficient catalysis is reported, except for the oxazaline based ligand complexes reported by Arnanz et al. These complexes are successfully employed in asymmetric hydrogenation. There are however no further reports of catalysis with these complexes. Concluding, this thesis showed that a general design motive for chiral NHC ligands is required for the expansion of applications of NHC ligands in asymmetric catalysis. Monodentate NHC ligands could not provide a general design motive because of difficulties with the definition of chiral space in these ligands. This thesis focussed on C2-symmetric di-NHC bidentate ligands because of the advantages of C2-symmetric ligands in reducing the number of diastereomeric transition states compared to non51 symmetric ligands. Bidentate ligands have shown to be able to define chiral space very well, and diNHC complexes of numerous metals have shown to have high (thermal) stabilities. Reports of syntheses of C2-symmetric di-NHC bidentate ligands have shown that two ligand design motives could be synthesised and modified easily. One of those ligand designs, the atropisomeric ligands reported by Shi, also showed to be applicable in a wide range of asymmetric catalyses, with various reports of excellent yields and ee’s. The catalysts were tolerant to electronic variations within the substrates leading to a considerable substrate scope for most of the reactions reported. The ligand design reported by Shi et al. is therefore a general and widely applicable ligand design for the synthesis of chiral NHC ligands. Because of the remarkable properties of N-heterocyclic carbenes and the strong ligand design motive reported, there is no doubt that further research into these ligands will provide the organometallic chemistry with an effective catalyst that uses a chiral N-heterocyclic carbene ligand. 52 7. References (1) Heitbaum, M.; Glorius, F.; Escher, I. Angew. Chem. Int. Ed. 2006, 45, 4732–62. (2) Van Leeuwen, P. W. N. M. Homogeneous Catalysis Understanding the Art; 4th ed.; Kluwer Academic Publishers: Dordrecht, 2004. (3) Herrmann, W. A.; Elison, M.; Fischer, J.; Kocher, C.; Artus, G. R. J. Angew. Chem. Int. Ed. 1995, 34, 2371–2374. (4) Herrmann, W. A.; Reisinger, C.; Spiegler, M. J. Organomet. Chem. 1998, 557, 93–96. (5) Böhm, V. P. W.; Weskamp, T.; Gstöttmayr, C. W. K.; Herrmann, W. A. Angew. Chem. Int. Ed. 2000, 39, 1998–2000. (6) Frenzel, U.; Weskamp, T.; Kohl, F. J.; Schattenmann, W. C.; Nuyken, O.; Herrmann, W. A. J. Organomet. Chem. 1999, 586, 263–265. (7) Scholl, M.; Trnka, T. M.; Morgan, J. P.; Grubbs, R. H. Tetrahedron Lett. 1999, 40, 2247–2250. (8) Coleman, A. W.; Hitchkock, P. B.; Lappert, M. F.; Maskell, R. K.; Müller, J. H. J. Organomet. Chem. 1983, 250, 9–14. (9) Tsutomu, K.; Sharpless, K. B. J. Am. Chem Soc. 1980, 5974–5976. (10) Noyori, R.; Ohkuma, T.; Kitamura, M.; Takaya, H.; Sayo, N.; Kumobayashi, H.; Akutagawa, S. J. Am. Chem Soc. 1987, 5856–5858. (11) Whitesell, J. K. Chem. Rev. 1989, 89, 1581–1590. (12) Berthod, M.; Mignani, G.; Woodward, G.; Lemaire, M. Chem. Rev. 2005, 105, 1801–36. (13) Öfele, K. J. Organomet. Chem. 1968, 12, 42–43. (14) Arduengo, A. J.; Harlow, R. L.; Kline, M. J. Am. Chem Soc. 1991, 113, 363–365. (15) Öfele, K.; Herrmann, W. A.; Scherer, W. J. Organomet. Chem. 1993, 459, 177–184. (16) Crabtree, R. H. Coord. Chem. Rev. 2013, 257, 755–766. (17) Donnelly, K. F.; Petronilho, A.; Albrecht, M. Chem. Comm. 2013, 49, 1145–59. (18) Nguyen, S. T.; Johnson, L. K.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem Soc. 1992, 2, 3974–3975. (19) Huang, J.; Stevens, E. D.; Nolan, S. P.; Petersen, J. L. J. Am. Chem Soc. 1999, 2674–2678. (20) Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–6. (21) Gusev, D. G. Organometallics 2009, 28, 6458–6461. 53 (22) Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–76. (23) Herrmann, W. A.; Köcher, C. Angew. Chem. Int. Ed. 1997, 36, 2162–2187. (24) Poyatos, M.; Mata, J. a; Peris, E. Chem. Rev. 2009, 109, 3677–707. (25) Jeletic, M. S.; Lowry, R. J.; Swails, J. M.; Ghiviriga, I.; Veige, A. S. J. Organomet. Chem. 2011, 696, 3127–3134. (26) Powell, M. T.; Hou, D.; Perry, M. C.; Cui, X.; Burgess, K. J. Am. Chem Soc. 2001, 8878–8879. (27) Seiders, T. J.; Ward, D. W.; Grubbs, R. H. Org. Lett. 2001, 3, 3225–8. (28) Wang, F.; Liu, L.; Wang, W.; Li, S.; Shi, M. Coord. Chem. Rev. 2012, 256, 804–853. (29) César, V.; Bellemin-Laponnaz, S.; Gade, L. H. Chem.Soc. Rev. 2004, 33, 619–36. (30) Herrmann, W. A. Angew. Chem. Int. Ed. 2002, 41, 1290–309. (31) Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B. Chem. Rev. 2009, 109, 3561–98. (32) Lappert, M. F. J. Organomet. Chem. 1988, 358, 185–214. (33) Duan, W.; Shi, M.; Rong, G. Chem. Comm. 2003, 2916–2917. (34) Liu, Z.; Shi, M. Tetrahedron: Asymmetry 2009, 20, 119–123. (35) Ma, G.-N.; Zhang, T.; Shi, M. Org. Lett. 2009, 11, 875–8. (36) Mata, J. A.; Poyatos, M.; Peris, E. Coord. Chem. Rev. 2007, 251, 841–859. (37) Liu, L.; Wang, F.; Shi, M. Organometallics 2009, 28, 4416–4420. (38) Chen, L.; Liu, Y.; Hou, G.; Song, H.; Zi, G. Inorg. Chem. Commun. 2013, 29, 141–144. (39) Song, H.; Fan, D.; Liu, Y.; Hou, G.; Zi, G. J. Organomet. Chem. 2013, 729, 40–45. (40) Veige, A. S.; Jeletic, M. S.; Lowry, R. J.; Abboud, K. A. Catalysts containing N-heterocyclic carbenes for enenatioselective synthesis, 2010. (41) Jeletic, M. S.; Ghiviriga, I.; Abboud, K. A.; Veige, A. S. Organometallics 2007, 5267–5270. (42) Mullick, A. B.; Jeletic, M. S.; Powers, A. R.; Ghiviriga, I.; Abboud, K. A.; Veige, A. S. Polyhedron 2013, 52, 810–819. (43) Jeletic, M. S.; Ghiviriga, I.; Abboud, K. A.; Veige, A. S. Dalt. Trans. 2010, 39, 6392–4. (44) Lowry, R. J.; Jan, M. T.; Abboud, K. A.; Ghiviriga, I.; Veige, A. S. Polyhedron 2010, 29, 553–563. 54 (45) Lowry, R. J. Rhodium and iridium complexes supported by chelating bis-N-heterocyclic carbene ligands: Design, Synthesis and Catalytic investigation, 2009. (46) Larrow, J. F.; Jacobsen, E. N.; Gao, Y.; Hong, Y.; Nie, X.; Zepp, C. M. J. Org. Chem. 1994, 1939– 1942. (47) Bonnet, L. G.; Douthwaite, R. E.; Hodgson, R. Organometallics 2003, 22, 4384–4386. (48) Gigler, P.; Bechlars, B.; Herrmann, W. A.; Kühn, F. E. J. Am. Chem Soc. 2011, 133, 1589–96. (49) Riederer, S. K. U.; Bechlars, B.; Herrmann, W. a; Kühn, F. E. Dalt. Trans. 2011, 40, 41–3. (50) Perry, M. C.; Cui, X.; Burgess, K. Tetrahedron: Asymmetry 2002, 13, 1969–1972. (51) Shigeng, G.; Tang, J.; Zhang, D.; Wang, Q.; Chen, Z.; Weng, L. J. Organomet. Chem. 2012, 700, 223–229. (52) Anezaki, S.; Yamaguchi, Y.; Asami, M. J. Am. Chem Soc. 2011, 696, 2399–2405. (53) Arnanz, A.; González-Arellano, C.; Juan, A.; Villaverde, G.; Corma, A.; Iglesias, M.; Sánchez, F. Chem. Comm. 2010, 46, 3001–3. (54) Marshall, C.; Ward, M. F.; Harrison, W. T. A. Tetrahedron Lett. 2004, 45, 5703–5706. (55) Riederer, S. K. U.; Bechlars, B.; Herrmann, W. A.; Kühn, F. E. Eur. J. Inorg. Chem. 2011, 249– 254. (56) Schneider, S. K.; Schwarz, J.; Frey, G. D.; Herdtweck, E.; Herrmann, W. a. J. Organomet. Chem. 2007, 692, 4560–4568. (57) Pfaltz, A.; Roseblade, S. J. Acc. Chem. Res. 2007, 1402–1411. (58) Perry, M. C.; Cui, X.; Powell, M. T.; Hou, D.-R.; Reibenspies, J. H.; Burgess, K. J. Am. Chem Soc. 2003, 125, 113–23. (59) Zhou, J.; Burgess, K. Angew. Chem. 2007, 119, 1147–1149. (60) Cui, X.; Ogle, J. W.; Burgess, K. Chem. Commun. 2005, 672–674. (61) Xu, Q.; Gu, X.; Liu, S.; Dou, Q.; Shi, M. J. Am. Chem Soc. 2007, 2240–2242. (62) Clavier, H.; Guillemin, J.; Mauduit, M. Chirality 2007, 476, 471–476. (63) Zhang, T.; Shi, M. Chem. Eur. J. 2008, 14, 3759–64. (64) Xu, Q.; Zhang, R.; Zhang, T.; Shi, M. J. Org. Chem. 2010, 75, 3935–7. (65) Liu, Z.; Shi, M. Tetrahedron 2010, 66, 2619–2623. (66) Liu, Z.; Shi, M. Organometallics 2010, 29, 2831–2834. 55 (67) Wang, W.; Zhang, T.; Shi, M. Organometallics 2009, 2640–2642. (68) Zhang, R.; Xu, Q.; Zhang, X.; Zhang, T.; Shi, M. Tetrahedron: Asymmetry 2010, 21, 1928–1935. (69) Duan, H.-F.; Xie, J.-H.; Qiao, X.-C.; Wang, L.-X.; Zhou, Q.-L. Angew. Chem. Int. Ed. 2008, 47, 4351–3. (70) Liu, Z.; Gu, P.; Shi, M. Chem. Eur. J. 2011, 17, 5796–9. (71) Hoffmann-Röder, A.; Krause, N. Angew. Chem. Int. Ed. 2004, 43, 1196–216. (72) Somei, M.; Yamada, F. Nat. Prod. Rep. 2004, 21, 278–311. (73) Zhang, R.; Wang, D.; Xu, Q.; Jiang, J.; Shi, M. Chin. J. Chem. 2012, 30, 1295–1304. (74) Hintermann, L.; Togni, A. Angew. Chem. Int. Ed. 2000, 39, 4359–4362. (75) Wang, W.; Wang, F.; Shi, M. Organometallics 2010, 29, 928–933. (76) Jacobsen, E. N.; Markó, I.; Mungall, W. S.; Schröder, G.; Sharpless, K. B. J. Am. Chem Soc. 1988, 1968–1970. (77) Wang, A.; Jiang, H.; Chen, H. J. Am. Chem Soc. 2009, 131, 3846–7. (78) Muñiz, K.; Hövelmann, C. H.; Streuff, J. J. Am. Chem Soc. 2008, 763–773. (79) Zhang, R.; Xu, Q.; Mei, L.; Li, S.; Shi, M. Tetrahedron 2012, 68, 3172–3178. 56