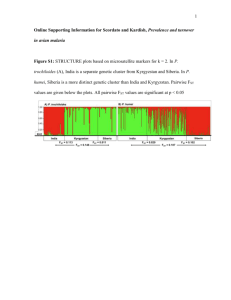
Supporting Information S1.¾Survey of previously published studies
advertisement

Supporting Information S1.Survey of previously published studies on Xenarthra and Phylogenetic analysis of previously published and newly generated DNA sequences. We used the ISI Web of Knowledge and Google Scholar online databases to look for previously published studies of Xenarthra. Phylogenetic analysis were performed including newly generated and previously published molecular data. All these evidences were discussed at both intra and interspecific levels and under the scope of four main biogeographic patterns already identified within the Neotropics (see introduction for further details). Therefore, not all available genetic studies on Xenarthra’ species were included in the present article. The search for previously published studies of Xenarthra was performed using a combination of terms including “Cingulata”, “Pilosa”, “Xenarthra”, “genetics”, “phylogeny”, “phylogeography”, “anteaters”, “armadillos”, “sloths”, “population”, and “genetic diversity”, and encompassed articles published up to June 2014. We analyzed previously published and newly generated molecular data from different xenarthran species in a molecular phylogenetic approach. All new data were generated from DNA samples used in Moraes-Barros et al. (2011), samples previously deposited in the Labec-IB/USP DNA and tissue collection (Brazilian Ministry of Environment-IBAMA, D.O.U. 28/200), or donated by different researchers (Supporting Information S2). The newly generated data were analyzed together with previously published sequences available in Genbank and encompassed DNA sequences of the mitochondrial gene cytochrome oxidase c subunit I (COI) from the sloths C. hoffmanni, B. torquatus and B. variegatus. DNA was extracted using the Invitrogen DNA purification kit (Forensic Charge Switch, Life technologies). COI segments were amplified by polymerase chain reaction (PCR) using the primers described in Lara-Ruiz et al. (2008). Amplification reactions (10 μl) consisted of 0.5μM of each primer, 2.5 mM MgCl2, 1 unit of Taq Platinum Polymerase (Invitrogen/Life technologies), and 0.8 mMdNTPs. Cycling conditions consisted of 94 °C for 7 min, 35 cycles of 93 °C for 35 s each, 54 °C for 40 s, 72 °C for 1 min, followed by a final extension at 72 °C for 10 min. Amplification products were purified by adding Exonuclease I (500 u/μl) and Shrimp Alkaline Phosphatase (100 u/μl), then maintained at 37 °C for 45 min and for 15 min at 80 °C. Sequencing was undertaken using 2 μl of the purified product, 0.5 μM of PCR primer and the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems) in a 20 μl final volume. The sequences obtained were analyzed and edited using 4 Peaks V. 1.7.2 software (© 2005, Mek and Tosj). The COI sequences comprised 612 base pairs and were aligned using Se-al 2.0a11 software (©1996-2002, Andrew Rambaut) in 2 data sets. The 1st contained new DNA sequences from 18 individuals representing all B. torquatus haplotypes described in Lara-Ruiz et al. (2008). Two DNA sequences were also used as outgroups, from C. hoffmanni (AMNH273184; Supporting Information S2) and T. tetradactyla (Genbank accession number AJ421450; Arnason et al. 2002). The 2nd data set contained 3 newly generated C. hoffmanni sequences (Supporting Information S2), the C. hoffmanni DNA sequences described in Steiner et al. (2010) for which geographical origin was known, and 2 DNA sequences, from B. torquatus (haplotype 3; Lara-Ruiz et al. 2008) and T. tetradactyla (Arnason et al. 2002), used as outgrups. All new DNA sequences are available from Genbank under accession numbers KP636540-KP636561. For B. variegatus the 2 individuals from the southern Atlantic forest shared the same haplotype. The 9 individuals from Panama also shared the same haplotype, so only 1 sequence is presented in the phylogeny (Fig. 2, main text). Phylogenetic relationships were reconstructed in Mega 5.0 software (Tamura et al. 2011) through neighbor-joining (NJ). The most appropriate model of DNA substitution for each data set was chosen according to the lowest BIC score (Bayesian Information Criterion). The robustness of neighbor-joining trees was determined by 10,000 bootstrap replications. Tamura-Nei (Tamura and Nei 1993) was the best model selected for both C. hoffmanni ( distribution of shape parameter G = 0.6; proportion of invariable sites I = 0.93) and Bradypus (I= 0.61) datasets. We also inferred phylogenies through a Bayesian approach by using Mr. Bayes 3.2.2 software (Ronquist et al., 2012). For both datasets we used only haplotypes and explored across the GTR model space in the Bayesian MCMC analysis (Huelsenbeck et al., 2004) to select an appropriate substitution model (GTRsubmodel [121324] for Choloepus and GTRsubmodel [123141] for Bradypus). Each MrBayes was run on 4 chains (temperature = 0.1) for 200,000 generations with trees sampled every 500 generations. The first 25% was discarded as burn in. We estimated divergence times for the main split events using as calibration points the split between B. torquatus and B. variegatus (14-11 mya) inferred by Moraes-Barros et al. (2011) and the split between Cholopeus and Bradypus (21-18 mya) estimated by Delsuc et al. (2004). We also implemented the average nucleotide substitution rate (0.006 ± 0.0005) observed for sloths mtDNA genes by Moraes-Barros et al. (2011) when estimating divergence times in Bayesian analyses. We first tested the data sets against the null hypothesis of equal evolutionary rates among lineages by performing Tajima's test (Tajima 1993), implemented in MEGA5 (Tamura et al. 2011) and through the stepping-stone (SS) method (Xie et al., 2011) implemented in Mr.Bayes 3.2.2 (Ronquist et al., 2012). The molecular clock hypothesis could not be rejected for both B. variegatus (Tajimas’s test: X2= 0.01, P = 0.92; SS: strict-clock model marginal likelihood Ln = -2,566.30 vs. non-clock model marginal likelihood Ln = -2,589.72) and C. hoffmanni (X2 = 0.10, P = 0.76; SS: strictclock model marginal likelihood Ln = -1,941.38 vs. non-clock model marginal likelihood Ln = -1,950.84) datasets. REFERENCES ARNASON, U. ET AL. 2002. Mammalian mitogenomic relationships and the root of the eutherian tree. Proceedings of the National Academy of Sciences U.S.A. 99:8151– 8156. RONQUIST, F. ET AL. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542. TAJIMA, F. 1993. Simple methods for testing molecular clock hypothesis. Genetics 135:599– 607. TAMURA, K., AND M. NEI. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution10:512–526. TAMURA, K., D. PETERSON, N. PETERSON, G. STECHER, M. NEI, AND S. KUMAR. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using Likelihood, Distance, and Parsimony methods. Molecular Biology and Evolution 28:2731–273. XIE, W., P. O. LEWIS, Y. FAN, L. KUO, AND M.-H. CHEN. 2011. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Systematic Biology 60:150–160.