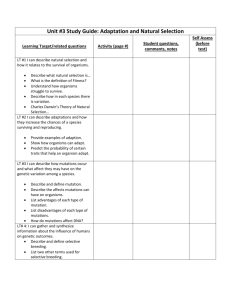
Garner Jacob Garner Dr. Ely BIOL 303 1 November 2013 Paternal
advertisement

Garner 1 Jacob Garner Dr. Ely BIOL 303 1 November 2013 Paternal Environmental Factors’ Impact on Offspring In the past when studying the health and development of fetuses and infants, the environmental and medical factors affecting the mother were the primary focus of study. This concentration made sense pragmatically, as a mother carries the fetus, sharing an environment during the gestation period. In the present, risks to an unborn child caused by maternal factors are fairly well known, such as an increased chance of birth defects (especially Down syndrome) as a mother ages and the correlation between maternal smoking and premature birth. Only recently have extensive studies been done on paternal factors that affect the health of a child, but such studies have shown that environmental factors affecting the father may be just as important as or perhaps more important than those affecting the mother at the time of conception. A study was performed by Dubrova et al. (2002) to validate a previously constructed hypothesis that radiation from the Chernobyl incident caused an increase in the germline mutation rate in the exposed populations. Although Dubrova et al. had performed a previous study in 1996, a difference in ethnicity between the group exposed to radiation and the control group was a source of confounding, making findings uncertain. In the 2002 study, DNA samples from both exposed and unexposed samples of the Belarus and Ukraine populations were used to analyze changes in the rate of germline mutations. Garner 2 Mutations of the germline are significant because the germline consists of cells that may be passed to offspring, gametes and gametocytes as opposed to somatic cells. Hence, these mutations are passed to the offspring without being expressed in the parents. Mutation rates in general are extremely low, and germline mutations are not an exception. In order to study the germline mutation rate, minisatellites were used. These sections of DNA are non-coding, repeating sequences varying from six to over a hundred base pairs in length. (They are greater in length than microsatellites as the prefix implies.) Since they are non-coding, the actual mutation of minisatellites does not have any significant impact on offspring, but the sites were used due to their high mutation rate, which provided more data. In the study, the minisatellite loci CEB1, CEB15, CEB25, CEB36, MS1, MS31, MS32, and B6.7 were used due to their high frequency of mutation as compared to that of other minisatellites. In order to find germline mutations, DNA was extracted from blood samples of parents from Ukraine and Belarus (some exposed to radiation from Chernobyl and some in control groups) and their children. The DNA was digested by the endonuclease AluI, size-fractionalized through agarose gel electrophoresis, and transferred to nylon membranes. Maternal and paternal germline mutations were observed in the form of minisatellite bands that were present in the offspring DNA samples, but were not present in the samples of the mother or the father. If an offspring sample contained a mutant band and a second band that matched a band in the mother’s sample, the germline mutation was paternal; similarly, if the second band matched a paternal band, the mutation was maternal. Garner 3 Figure 1 (Linschotten et al. 2013) On the left, no mutation is present. Each of the child’s bands matches a parental band. On the right, the arrow denotes a paternal germline mutation. One of the child’s bands matches the mothers, making the one matching neither (the mutation) paternal. The expected increase in germline mutations in response to radiation was observed but only for paternal mutations. Irradiated fathers showed a statistically significant increase in mutation rate by a factor of 1.6 compared to their non-irradiated counterparts while there was no statistically significant increase in mutation rate in irradiated mothers (Dubrova et al. 2002). In this sense, the study succeeded in demonstrating an increased mutation rate linked to radiation exposure but also presented a difference in the frequency of maternal and paternal minisatellite germline mutations. It was likely the timing of oogenesis and spermatogenesis caused this difference (Dubrova et al. 2002). Spermatogenesis is an ongoing process beginning at puberty while oogenesis is completed in the embryonic stage of development. Hence, the gametes of the mothers exposed to radiation had already formed prior to the incident, preventing mutation while the sperm would have been formed by DNA replication and meiosis after the radiation had occurred. However, this explanation is merely a hypothesis; the data only show an increase in paternal germline mutations as a result to radiation exposure and no accompanying increase in maternal germline mutations. Garner 4 Figure 2 (Dubrova et al. 2002) The graph represents the significant difference in mutation rate between exposed and unexposed fathers (left) and lack of significant difference between exposed and unexposed mothers (right). One potential shortcoming in the post-Chernobyl study was a difference in the average age of fathers in the irradiated and control groups; the mean age of fathers in the control group was 25.6 while the mean in the irradiated group was 26.8 (Dubrova et al. 2002). Thus, paternal age is a confounding variable, but any extreme effect of age was ruled out due to significantly higher mutation rates in exposed fathers in the same age group as controls. Notably, there was a slight positive correlation between age and mutation rate in the group of Ukrainian fathers exposed to radiation as seen in Figure 3. Figure 3 (Dubrova et al. 2002) In the context of the study, the graph shows a higher mutation rate among irradiated fathers, but a slight upward trend between age and mutation rate is also displayed. Garner 5 The trend of mutation rate increasing with age in the post-Chernobyl study was likely not coincidental. While the effects of age on mutation rate were not as severe as the effects of radiation, they did exist. Paternal age was shown to positively correlate with the rate of de novo mutations in a study by Kong et al. (2012). In the study, 78 triads of mothers, fathers, and children with autism spectrum disorder or schizophrenia had their genomes sequenced and examined for de novo SNP mutations to discover any correlation with paternal age. (De novo mutations originating in a gamete.) A difficulty in finding a relationship between the father’s age and the rate of SNP mutations was caused by a correlation between the ages of the parents. (Understandably, an older father tended to have an older partner.) In five cases, the offspring having the de novo mutation also had a child, allowing certain determination of the mutation’s origin. If the second-generation offspring inherited the paternal haplotype and had the mutation, it was a paternal de novo mutation, but if the mutation was not present, it was a maternal mutation; the same logic applies if the maternal haplotype was inherited. In the sample of five, the mean number of paternal mutations was 55.4 while the mean of maternal mutations was 14.2. More notably, the paternal variance was 428.8, much larger than the maternal variance of 48.7 (Kong et al. 2012). (The variance is the square of the standard deviation.) These data were used to show that the fathers were more responsible for the increased rate of de novo mutations as parents aged. In a majority of cases, however, no grandchild existed, necessitating the use of statistical methods to differentiate between the effects of maternal and paternal age. Hence, a multiple regression was used, revealing the father’s age was highly significant while the mother’s age was not (Kong et al. 2012). Therefore, the increase in mutation Garner 6 rate with age was nearly exclusively paternal. As shown in Figure 4, three potential relationships were found between paternal age and the number of de novo mutations: one linear, one exponential factoring in maternal and paternal ages, and one exponential assuming the maternal mutation rate is constant. While more data would be required from fathers over the age of forty to find the most accurate model, all models indicated paternal age accounted for over 95% of variation in de novo mutation rate. Figure 4 (Kong et al. 2012) Three possible models relating paternal age to mutation rate are shown. Despite a lack of data at the upper end to determine which model is accurate, a positive correlation is apparent. The two aforementioned studies focused on specific variables (radiation exposure and age) to find their effects on the paternal mutation rate. This approach showed the advantage of finding direct associations with few sources of confounding, but inversely, correlations with mutation rate could only be drawn regarding a single variable. Linschotten et al. (2013) performed an analysis of the effects general paternal lifestyle factors on germline mutation rates. Similarly to the 2002 study of Dubrova et al., minisatellite germline mutations were identified by DNA that was digested by AluI being electrophoresed and compared among triads of fathers, mothers, and children. However, only two loci, CEB1 and B6.7, were analyzed. Lifestyle information, including age, weight, height, education, employment, income, smoking status, and medical factors and Garner 7 risks, were collected from MoBa, the organization from which the DNA samples were obtained. After all the mutations had been discovered, the families were separated into a group with paternal mutations and a group without. Using a Fisher’s exact test, correlations were sought between each of the variables in the MoBa questionnaire and the presence of mutations. No significant relationships were discover at the B6.7 locus while at the CEB1 locus, relationships were found involving smoking and income; smoking fathers were more likely to pass mutations to their children via the germline compared to non-smokers as were fathers in the lower income range (earning below approximately seventy thousand USD yearly) (Linschotten et al. 2013). Interestingly, the mean age of the two groups did not significantly differ. A likely shortcoming of the study was a small samples size. Seventy-eight families were observed, but only eight paternal mutations were found at the CEB1 locus while only five were found at the B6.7 locus (Linschotten et al. 2013). A Fisher’s exact test is relatively accurate for smaller sample sizes, but given the number of mutations and categories examined, there was a large potential for false positives and undetected correlations. Although a correlation between smoking and mutation rate is not unreasonable, it was unusual that income should be one of the observed factors affecting mutation rate. Income could be used as a measure of overall health (i.e. a higher income allows one to afford a more healthful lifestyle), but no relationship was found among other health factors. Due to the small sample size, further research is necessary to validate the results. In all of the previously mentioned studies, there was a commonality in findings: an Garner 8 association between some factor affecting a father and mutation rate. This allowed for speculation about what effects these factors may have on a child’s health. For example, in the study of Kong et al. (2012), it was speculated that an increase in de novo mutations associated with an increase in paternal age would increase the probability of a mutation triggering either autism or schizophrenia. Despite the hypothesis, since nearly all the offspring had one of the two diseases, no correlation could be found. In all cases, the mechanism by which mutations could cause harm to a child was not observed, and thus the implications of the mutations were not certain. A study by Deng et al. (2013) was conducted with the intent to establish an association between paternal smoking and congenital heart defects (CHD) in offspring. Data regarding paternal smoking habits and health and lifestyle of the mother were collected from mothers of fetuses or infants with various types of CHD as well as a control group of mothers with healthy infants. The group reporting paternal smoking was further stratified into groups based on frequency of smoking (one to nine, ten to nineteen, or twenty or more cigarettes daily) as well as whether the father smoked in close proximity to the mother during pregnancy. After chi-squared testing and statistical methods to account for confounding, it was seen that paternal smoking was associated with an increased risk of various (but not all) types of heart defects with the hypothesized cause being carcinogens from cigarette smoke impacting spermatogenesis (Deng et al. 2013). Additionally, the impact of paternal age on rate of birth defects was analyzed by Yang et al. (2007). Data were harvested from the 1999-2000 national linked birth/infant mortality database provided by the CDC, which included information on parents’ health, Garner 9 lifestyle, and medical complications. Confounding was a serious issue in the study, especially due to the correlation in paternal and maternal age. Logistic regression was used to model the correlation between paternal age and chance of birth defects, and statistical software was used to adjust for confounding variables including maternal age, race, smoking, and alcohol use. A slight but significant elevation in odds of birth defects was found both when fathers were younger than 25 or older than 34 with the greatest risk associated with a father over 50 (Yang et al. 2007). Due to the various sources of confounding, it was concluded that paternal age had a small effect on the likelihood of birth defects. The results of various studies have shown that environmental and health factors affecting a father at the time of conception can cause mutations in offspring (Dubrova et al. 2002; Kong et al. 2012; Linschotten et al. 2013). Although the strength of various correlation and conflicting results leave the pertinence of individual factors in question, it is likely that factors such as paternal age, smoking, and exposure to radiation elevate the rate of mutations passed to offspring via the germline. Other studies have found that such factors correlate to birth defects in progeny (Deng et al. 2013; Yang et al. 2007). However, the genetic mechanism of the birth defects is more often hypothesized than studied. Due to the lack of information of how paternal germline mutations may affect offspring and what other factors may cause them, the connection between a father’s health prior to conception and the health of his children requires further exploration. Garner 10 Literature Cited: Deng, K., Liu, Z., Lin, Y., Mu, D., Chen, X., Li, J., Li, N., Deng, Y., Li, X., Wang, Y., Li, S. and Zhu, J. 2013. Periconceptional paternal smoking and the risk of congenital heart defects: A case-control study. Birth Defects Research Part A: Clinical and Molecular Teratology. 97: 210–216. http://onlinelibrary.wiley.com/store/10.1002/bdra.23128/asset/bdra23128.pdf?v=1&t= hnfdg38u&s=8ef619f10ea8f245ee061b533e2aa4d999496bc2 Dubrova YE, Grant G, Chumak AA, Stezhka VA, Karakasian AN. 2002. Elevated minisatellite mutation rate in the post-Chernobyl families from Ukraine. Am J Hum Genet 2002. 71: 801–809. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC378537/pdf/AJHGv71p801.pdf Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., Gudjonsson, S. A., Sigurdsson, A., Jonasdottir, A., Jonasdottir, A., Wong, W. S., Sigurdsson, G., Walters, G. B., Steinberg, S., Helgason, H., Thorleifsson, G., Gudbjartsson, D. F., Helgason, A., Magnusson, O. T., Thorsteinsdottir, U., and Stefansson, K. 2012. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 23: 471–475 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3548427/pdf/nihms391279.pdf Linschooten JO, Verhofstad N, Gutzkow K, Olsen AK, Yauk C, Oligschläger Y, Brunborg G, van Schooten FJ, Godschalk RW. 2013. Paternal lifestyle as a potential source of germline mutations transmitted to offspring. FASEB J. 27:2873–2879 http://www.fasebj.org/content/27/7/2873.full.pdf+html Yang Q, Wen SW, Leader A, Chen XK, Lipson J, Walker M. 2007. Paternal age and birth defects: how strong is the association? Hum Reprod 2007:22:696-701. http://humrep.oxfordjournals.org/content/22/3/696.full.pdf+html