mec13465-sup-0003-SupplementaryfileS3
advertisement

Supplementary Material online
Supplementary file S3. Bayesian estimation of mean mutation
rates at microsatellite loci from meiosis data and rates of homoplasy.
Homoplasy estimates
Sensitivity to prior information
Scripts in R
Homoplasy estimates
For each simulation and each locus, we selected a candidate allele for mutation by drawing 1)
one of the 26 families, with a probability equal to the frequency of offspring analysed in the
family, then 2) one of the four parental alleles of the family within the real genotype dataset
and with equiprobability. We then simulated a mutation event (see below) and recorded
whether the new allele variant was identical in state to one of the four parental alleles. The
number of homoplasic mutations in these simulations divided by the number of simulations
gave hi.
In order to simulate mutations in the repeat region of microsatellite loci, we used a
generalized stepwise mutation model (GSM; Zhivotovsky et al. 1997), in which the change in
the number of repeat units is a geometric random variable with probability of success p. We
followed standard parameterization of the GSM model (Estoup et al. 2001; Cornuet et al.
2008) with:
-
The mean parameter of the geometric distribution of the length in number of repeats of
mutation events (mean p) equaled to 0.78;
-
The parameters p for individual microsatellites were drawn from a gamma distribution
centered on mean p and with a shape of 2 (and only values in the range 0.01-0.99 were
retained).
-
Since constraints on allele size may often occur and favor size homoplasy (reviewed in
Estoup et al. 2002), we imposed range constraints with reflecting boundaries on the
mutation process (see main text and Range in table 2), while in-range mutations occur
symmetrically in each direction (Feldman et al. 1997).
The probability of undetected mutation averaged 11% over microsatellite loci, with
values from about 1% to 28%. It depended on the distribution of allele sizes at the loci in the
parental population. A low allelic size variance at microsatellite loci explained high rates of
false-negatives (linear model; t21=-2.68; P=0.014; see supplementary figure S3-1). This effect
was however moderate in our data, with a mean of allelic size variance of 35 at anonymous
microsatellites and 21 at expressed microsatellites (see also V in table 1 in the main
document).
The fraction of mutation events that reflected to the size range limits did not explain
significantly the differences in false-negative rates between markers, though the relation
seems positive (t21=1.24; P=0.228; see supplementary figure S3-2). This was because this
fraction was generally low, with a mean of 3.2% and a range from 0.5% to 10% (see also b in
table 1 in the main document).
The few loci concerned by effects of size constraints were those for which some
parental alleles were by random at the limit of the range (e.g. SgM41 and SgM87; see
supplementary figure S3-3) or those, rare, with a small range of continuous allele sizes (e.g.
diEST6 and diEST12; see also table 1 in main document).
Figure S3-1. Homoplasy rates (h) as a function of allelic size variance (V).
Figure S3-2. Homoplasy rates (h) as a function of the rate of size constraints (b).
Figure S3-3. Rates of size constraints (b) as a function of the minimum distance of parental
alleles to size limits (dlim).
References
Cornuet J-M, Santos F, Beaumont MA et al. (2008) Inferring population history with
DIYABC, a user-friendly approach to Approximate Bayesian Computation. Bioinformatics,
24, 2713–2719.
Estoup A, Jarne P, Cornuet J-M (2002) Homoplasy and mutation model at
microsatellite loci and their consequence for population genetics analysis. Molecular Ecology,
11, 1591-1604.
Estoup A, Wilson IJ, Sullivan C, Cornuet J-M, Moritz C. (2001) Inferring population
history from microsatellite and enzyme data in serially introduced cane toads, Bufo marinus.
Genetics, 159, 1671–1687.
Feldman MW, Bergman A, Pollock DD, Goldstein DB (1997) Microsatellite genetic
distances with range constraints: analytic description and problems of estimation. Genetics
145: 207–216
Zhivotovsky LA, Feldman MW, Grishechkin SA(1997) Biased mutations and
microsatellite variation. Molecular Biology and Evolution, 14, 926–933.
Sensitivity to prior information
Sensitivity analyses to prior values for the mean rate of mutation show that considering a
uniform distribution result in a 30% increase in the posterior mean (fig. S3-4; and point
estimate values in table S3-1). This upward trend is explained by a prior uniform assumption
that represented primarily the upper log-interval (i.e. 1e-4 to 1e-3; fig. S3-4) and further
strengthens our conclusions. In addition, setting priors for the shapes of the gamma
distribution to different fixed values would not change posterior point estimates but
confidence intervals (which would decrease with higher parameter values ; table S3-1).
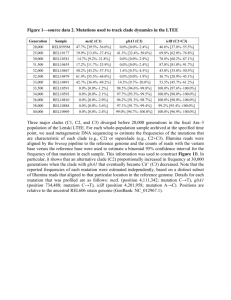
Table S3-1. Sensitivity of point estimates of mean mutation rates (μ) in S. gregaria
dinucleotide microsatellite loci to prior information.
Prior distribution
µ ~ LogU[1e-6;1e-3]
Origin
Mean
Mode
Median
90% CI
U
2.08e-4
1.35e-4
1. 82e-4
7.6e-5; 4.28e-4
T
9.1e-5
5.3e-5
7.4e-5
2.0e-5; 2.18e-4
U
1.96e-4
1.57e-4
1. 80e-4
8.1e-5; 3.66e-4
T
8.6e-5
5.3e-5
7.4e-5
2.2e-5; 1.92e-4
U
2.32e-4
1.32e-4
1. 89e-4
6.8e-5; 5.56e-4
T
1.03e-4
4.5e-5
7.7e-5
1.9e-5; 2.74e-4
U
2.72e-4
1.81e-4
2.39e-4
1.00e-4; 5.63e-4
T
1.41e-4
8.8e-5
1.18e-4
3.7e-5; 3.24e-4
α = 0.7
µ ~ LogU[1e-6;1e-3]
α = 1.4
µ ~ LogU[1e-6;1e-3]
α = 0.35
µ ~ U[1e-6;1e-3]
α = 0.7
Origin: untranscribed (U) or transcribed (T).
Figure S3-4. Sensitivity of posterior probability densities of mean mutation rates to a uniform
prior distribution.
Scripts in R
Script 1. Estimation of homoplasy (simulations)
########### DESCRIPTION ################
# Script R - microsatellite mutation rate in desert locust – estimation
of homoplasy
########### DATA ######################
# genotypes of parents
dataf<-read.table("Parentalgenotypes_DRYAD.txt", header = TRUE)
dataf<-dataf[,-(c(1,2))] # to keep allelic states only
nmarkers<-((ncol(dataf))/2)
nfamily<-(nrow(dataf)/2)
# vectors of size constraints
minf<c(188,168,168,106,211,254,189,199,160,149,326,197,113,218,156,169,113,2
33,205,247,250,157,247,234)
maxf<c(286,341,386,254,435,396,479,275,262,223,424,403,157,294,183,295,213,2
67,311,397,406,215,297,334)
# vector of offspring number per family (weight) to create
# a vector of family ID per offspring (familyf)
weight<c(12,76,144,102,59,104,44,112,104,87,8,56,121,56,12,8,32,15,4,55,116,48
,107,104,36,124)
familyf<-vector(length=sum(weight),mode="integer")
j<-1
for(i in 1:nfamily) {
familyf[j:(j+weight[i]-1)]<-i
j<-j+weight[i]
}
# simulation number
nrep<-100000
# vector of the number of mutations out of size range
# (over the nrep simulations) for each marker - initialization
limit<-vector(length=nmarkers,mode="integer")
# vector of the number of homoplasic mutations +
# mutations out of size range (over the nrep simulations)
# for each marker - initialization
count<-vector(length=nmarkers,mode="integer")
########### SIMULATIONS ######################
for(iter in 1:nmarkers) # for each marker
{
for(rep in 1:nrep) # for each rep
{
## DRAW A MUTANT ALLELE
index<-familyf[sample(1:sum(weight),1)] # draw a family (random)
alleles_maman<-c(dataf[(index*2)-1,(iter*2)-1],dataf[(index*2)1,(iter*2)]) # register mother alleles
alleles_papa<-c(dataf[(index*2),(iter*2)-1],dataf[(index*2),(iter*2)])
# register father alleles
x<-sample(alleles_maman,4,replace=T) # draw 4 times a mother allele
# (random)
y<-sample(alleles_papa,4,replace=T) # draw 4 times a father allele
# (random)
z<-c(x,y) # constitute the allelic pool of 4 offspring
real<-runif(1,0,1)
state<-ifelse (real < 0.5, z[1],z[5]) # draw a mutant allele in either
# mother or father (first allele)
## SIMULATE THE MUTATION EVENT UNDER A GSM MODEL
# draw the number of repeat change, which forms a geometric random
# variable with probability of success p
# draw, first, the microsatellite-specific p values (<0.99) from a
# gamma distribution with a mean of 0.78
p<-rgamma(1,2,2/0.78)
while((p>0.99) | (p<0.01)) p<-rgamma(1,2,2/0.78)
deltamu<-rgeom(1,p)
while(deltamu==0) deltamu<-rgeom(1,p)
# determine the new state, either a deletion or an insertion
# (in size for dinucleotide microsatellites)
real<-runif(1,0,1)
new_state<-ifelse ((real<0.5),(state-(deltamu*2)),(state+(deltamu*2)))
# apply size constraints (back to ancestral state - would be remove
# from the total count)
if ((new_state > maxf[iter]) | (new_state < minf[iter])) {
new_state<-state
limit[iter]<-limit[iter]+1
}
# detect if the new state is homoplasic
detect<-TRUE
for (i in 1: 2) {
if ((alleles_maman[i]==new_state) | (alleles_papa[i]==new_state))
{
detect<-FALSE
break
}
}
if (detect==FALSE) {count[iter]<-count[iter]+1}
}
}
#end per rep
#end per marker
# save in an excel output file
namesf<-colnames(dataf)
listf<-c(1:(nmarkers*2))
is.odd <- function(x) x %% 2 != 0
list.odd<-listf[is.odd(listf)]
marker.names<-namesf[list.odd]
out<-data.frame(marker.names,(count-limit)/nrep,limit/nrep)
write.table(out,paste("homoplasy.xls",sep=""),sep="\t",col.names=c("mar
kers","false_negative","out_of_range"),row.names=FALSE)
Script 2. Estimation of mutation rates (Bayesian model)
This script requires BRugs package and OpenBugs software version 3.2.2 for performing
Gibbs sampling-based Bayesian inference.
########### DESCRIPTION ################
# Script R - microsatellite mutation rate in desert locust – estimation
# of mutation rates
########### MODEL ######################
# example of model structure for homoplasy consideration in
# hierarchical bayesian inference:
model<-function(){
#likelihood:
for(i in 1:Nmarker){
mutation[i]~dbin(eta[i],M)
eta[i]<-mu[i]-mu[i]*homoplasy[i]
mu[i]~dgamma(alpha, beta[markertype[i]] )
}
#prior:
alpha <-0.7 #fixed
for(k in 1:2){
beta[k]<-alpha/pop.mean[k]
#to obtain a log-uniform prior on mean
#mutation rate (pop.mean):
pop[k] ~ dunif(-13.81551, -6.907755)
pop.mean[k] <- exp(pop[k])
}
}
########### DATA ######################
# data to include for posterior estimation:
M<-3492
# meiosis number
Nmarker<-24 # number of markers
# number of mutations observed per marker:
mutation<-c(0,0,1,0,2,0,0,0,0,2,1,1,0,0,0,0,0,0,0,0,3,0,0,0)
# homoplasic effect estimated per marker:
homoplasy<c(0.08657231,0.08618444,0.13108261,0.01315332,0.01245538,0.03561121,0.1
9983249,0.08804119
,0.09109291,0.16683753,0.05481716,0.10152357,0.20778627,0.02080252,0.08
568421,0.05016842
,0.13090824,0.20106041,0.08301236,0.05020572,0.03281510,0.28455812,0.25
869771,0.17607620)
# marker types:
markertype<-c(rep(2,12),rep(1,12))#1=transcribed, 2=untranscribed
########## BRUGS COMMANDS ##############
# necessary R package:
require(BRugs)
filenameD<- "data.txt"
# create data file (txt):
bugsData(list(Nmarker=Nmarker,M=M,mutation=mutation,markertype=markerty
pe,homoplasy=homoplasy),fileName = filenameD, digits = 5)
filenameM <- "model.txt"
writeModel(model, filenameM)
# create model file (txt)
modelCheck(filenameM)
# check model file
modelData(filenameD)
# read data file
modelCompile(numChains=3)
# compile model with 3 chains
modelGenInits()
# generate random initial values from priors
modelUpdate(100000,thin=1)
# burn-in phase (~30 seconds)
samplesSet(c("mu","pop.mean")) # set observer for nodes of interest
modelUpdate(10000,thin=10)
# sampling phase (~30 seconds)
sast<-samplesStats("*")
# get the chains' statistics for nodes
of interest
saD<-samplesDensity("*",mfrow=c(3,3)) # plot the densities for nodes of
# interest
sH<-samplesHistory("*",mfrow=c(3,3)) # plot the history (chains) for
# nodes of interest
print(sast)
# show (in R console/output) the chains' statistics