Chapter 9 Chemical Bonding
advertisement

Chapter 9 Molecular Geometry and Bonding Theories
Molecular geometry refers to the three-dimensional arrangement of atoms in a molecule.
A number of properties of molecules (e.g., physical properties such a melting points and
boiling points) and chemical properties are affected by the molecules’ arrangement of
atoms in space.
NOTE: LEWIS STRUCTURES CAN’T BE USED TO PREDICT GEOMETRY
A very simple theory advanced by Gillespie in Montreal tells us that the repulsion between
electron pairs (both bonding and non-bonding) helps account for the arrangement of atoms
in molecules.
Note electrons are negatively charged, they want to occupy positions such that electron –
electron interactions are minimised as much as possible.
Valence Shell Electron-Pair Repulsion Model
(1)treat double and triple bonds as single bonds
(2)resonance structure - apply VSERR to any of them
Formal charges are use omitted
Central Atom (designated A) no lone pairs (for simplicity, the lone pairs on the
surrounding atoms are omitted).
Summary of Rules for VSEPR
(1)
Draw Lewis Structure.
(2)
Identify the central atom.
(3)
Count number of lone pairs and bonding pairs (note that double and triple bonds are
counted as 1).
(4)
Obtain the ideal geometry form VSEPR tables (9.1-9.3 in Brown, Lemay, and Bursten).
Check for lone pair repulsions to correct the ideal geometry. Also check for connections
due to the presence of multiple bonds (double and triple bonds).
AB2
the Cl-Be-Cl is 180 .
Cl
Be
Cl
2
AB3 planar molecule (in one plane) Cl-B-Cl = 120; the electron pair repulsions are
minimized.
Lewis Structure
Cl
B
Cl
Cl
AB4
VSEPR
Structure
Structure
Cl
B Cl
Cl
Classic examples are methane (CH4) and carbon tetrachloride (CCl4).
Lewis Structure
H
H
C
H
H
VSEPR
Structure
Structure
H
H C H
H
VSEPR predicts a tetrahedral structure (i.e., HCH 109.5)
Lewis Structure
Cl
Cl P Cl
Cl
Cl
AB5
e.g., PCl5
VSEPR
Structure
Structure
Cl
Cl
Cl P Cl
Cl
VSEPR predicts a trigonal bipyramid
Note: Cleq P Cleq = 120 (eq – equatorial chlorine atoms)
Clax P Cleq = 90 (ax – axial chlorine atoms)
Clax P Clax = 180.
3
AB6
SF6 octahedral arrangement predicted by VSEPR
VSEPR
Structure
Structure
F
F
F
S
F
F
F
Lewis Structure
F F F
S
F F F
Molecules in which the central atom has one (or more) lone pairs.
We have to look at the strength of the e—e- pair interactions.
lone pair - lone pair > lone pair - bonding pair = > bp-bp
Reason lone pair electrons occupy more space and experience a greater repulsion from
neighbouring lone pairs and bonding pairs. Bonding pairs are localized between nuclei
(i.e. in the bond) and are fairly well held by the attractive e—nuclear forces. Lone pairs are
localized on atoms.
Examples
AB2E
SO2
Lewis Structure
O S O
VSEPR
Structure
Structure
S
O
O
For VSEPR, we treat the double bonds as single bonds. This gives us 3 e- pairs around the
S central atom (2 bonding + a lone pair) VSEPR predicts that the electron pair geometry
will be trigonal planar.
Note: the OSO angle is <120 (119.5) due to lone pair – bonding pair interactions.
AB3E NH3
Lewis Structure
H N H
H
VSEPR
Structure
StructureN
H
H
H
4
Around central N atom, 4 e pairs (1 lone pair and 3 bonding pairs) VSEPR predicts that
the electron pair geometry will be tetrahedral.
-
AB2E2 H2O
H-O-H
Lewis
Note around O atom, 2 bonding pairs and 2 lone pairs. the electron pair geometry will
again be a tetrahedral, but the two lone pairs interact with themselves as well as the bonding
pair.
VSEPR
H
O
H
“ideal” HOH = 109.5, actual
104.5
AB4E SF4
Lewis
F
F
S
F
F
From VSEPR theory, there are 5 e- pairs around the central S-atom the electron pair
geometry will be trigonal bipyramidal.
Where does the lone pair go?
F
S
F
F F
1
F F
S F
F
2
5
Which one has the greatest repulsions?
Structure [1]
2 L.p. - B.p.’s at 90
2 L.p. - B.p.’s at 120
Structure [2]
3 L.p. - B.p.’s at 90
1 L.p. - B.p. at 180
less repulsion here
Structure [1] is the observed structure (a “see-saw” shape)
Geometry for molecules with more than 1 central atom
Example:
C2H6
There are two central C atoms.
C atom (1) 4 bonding pairs.
C atom (2) the same
the geometry around both atoms is the same (tetrahedral)
H
H
C
H
H
C H
H
We predict the geometry around each atom is 109.5 ( HCH and HCC).
Dipole Moments
Some molecules have charge separation. Pick a covalently bonded molecule like HF
+
H-F
arrow indicates the e- distribution is uneven and towards the F.
quantitative measure dipole moment
=Q*r
Q = charges; r = distance between them
homonuclear diatomics no dipole moment (O2, F2, Cl2, etc)
heteronuclear diatomics polar molecules (i.e., there is a charge separation generally
possess a dipole moment)
-expressed in Debye units (D)
1 D = 3.33 * 10-30 c.m. (Coulomb meter)
6
Triatomic molecules (and greater). Must look at the net effect of all the bond dipoles.
e.g., CO2
+
+
O=C=O
each C=O bond has a charge separation.
Charge separation in CO2 is in equal, but in opposite directions
no dipole moment.
e.g.
CCl4
Cl
Cl
C Cl
Cl
Tetrahedral molecule, each bond has a charge separation (a bond moment)
Note CCl4 is a regular tetrahedron all the individual bond dipoles cancel no
resultant dipole moment.
Valence Bond Theory and Hybridisation
Valence bond theory permits the description of the covalent bonding and structure in
molecules. Assumes that electron in a molecule occupy the atomic orbitals of individual
atoms. The covalent bond results from the overlap of the atomic orbitals on the individual
atoms.
Examples
H2 each H atom contributes a 1s1 electron to the formation of the covalent bond.
H-H
Bonding description
1s(H1) – 1s(H2)
type orbital.
Cl2 each Cl atom contributes a 3pz electron to the formation of the covalent bond.
Cl-Cl
Bonding description 3pz (Cl 1) – 3pz (Cl 2) type orbital.
These orbitals are sigma () type bonds they have cylindrical symmetry with respect to
an imaginary line joining the nuclei of the two bonded atoms.
Note that in many cases, simply overlapping atomic orbitals on adjacent atoms may not
permit us to rationalise both the geometry and the number and type of bonds present in
molecules. In many cases, the bonding and geometry in polyatomic moles may be
explained in terms of the formation of hybrid atomic orbitals on the central atom and the
7
subsequent overlap of the hybrid atomic orbitals with the appropriate half-filled atomic
orbital on the terminal atoms.
Definition
Hybridisation of Atomic Orbitals - the mixing of atomic orbitals in a central atom to generate
a new set of equivalent atomic orbitals. Note that these new atomic orbitals are of different
energy and different geometry that the atomic orbitals used to construct them.
e.g. sp3 hybridisation good example CH4.
We mix 3 “pure” p orbitals and a “pure” s orbital to form a “hybrid” or mixed orbital sp3.
e.g., for the C central atom
Orbital diagram for the valence shell
1s2
2s2
2p2
Imagine promoting an e-
2s1
2p3
We then “mix” to form four equivalent orbitals, the sp3 hybrid orbitals.
4 sp3 hybrid orbitals
in CH4, the carbon sp3 orbitals (4 of them) overlap with the H 1s orbitals to form the CH bond.
8
Extend to ethane C2H6
CH3CH3
H H
H
C C H
H H
Bond overlaps
[sp3 (C 1 ) – 1s (H) ] x 6
type
[sp3 (C 1 ) - sp3 (C 2) ] x 1
type
The formation of hybrid orbitals in C permits a greater overlap of the orbital greater
overlap means stronger bonding.
sp2 hybridisation
Look at ethene C2H4. The central carbon atoms each have a double bond and 2 single
bonds. Only 3 bonds are required for each carbon in the C2H4 molecule. Also, since each
central atom is an AB2 system, the bonding picture must be consistent with VSEPR theory.
we mix the 2s1 and “2” 2p orbitals and form 3 sp2 hybrids.
Again the total # of hybrid AO’s = total # of atomic orbitals used.
Note that again we start with the orbital diagram for C
2s2
Promote the electron.
2p2
2s1
2p3
But this time, we only need three equivalent types of orbitals, with the proper geometry.
Therefore, we mix or hybridise the s orbital and only two p orbitals to form three new sp2
hybrid atomic orbitals.
sp2 hybrid orbitals + an unhybridised p orbital
9
Bond overlaps
[sp (C 1 ) – 1s (H) ] x 4
type
[sp2 (C 1 ) – sp2 (C 2) ] x 1
type
2
What about the unhybridised p orbitals each containing an unpaired electron? We simply
overlap the two parallel 2pz orbitals (a -orbital is formed).
type
[2pz (C 1 ) – 2pz (C 2) ]
Bond angles HCH = HCC 120. Note that the bond is perpendicular to the plane
containing the molecule.
sp Hybridisation
Look at acetylene (ethyne)
H-CC-H
The carbon atoms each have a triple bond and a single bond. Only 2 bonds are required for
each carbon in the C2H2 molecule. Also, since each central atom is an AB system, the
bonding picture must be consistent with VSEPR theory (i.e., linear geometry).
we mix the 2s1 and “1” 2p orbital and form 2 sp hybrids.
Again the total # of hybrid AO’s = total # of atomic orbitals used.
Note that again we start with the orbital diagram for C
2s2
2p2
Promote the electron
2s1
2p3
We mix (hybridise) the s orbital and only a single p orbital to form two new sp hybrid
atomic orbitals.
sp2
H-CC-H
Bond overlaps
2py, 2pz
[sp (C ) – 1s (H) ] x 2
type
10
type
type
type
[sp (C 1 ) – sp (C 2) ]
[2py (C 1 ) – 2py (C 2) ]
[2pz (C 1 ) – 2pz (C 2) ]
Notes for Understanding Hybridisation
1. Applied to atoms in molecules only
2. Number hybrid orbitals = number of atomic orbitals used to make them
3. Hybrid orbitals have different energies and shapes from the atomic orbitals from which
they were made.
4. Hybridisation requires energy for the promotion of the electron and the mixing of the
orbitals energy is offset by bond formation.
spd Hybridisation
Explains the “expanded octets” of elements > 14. A good example is PCl5.
There are five bonds between P and Cl (all type bonds). How doe we ratioanlize the
bonding picture in this molecule (and other molecules with expanded valence shells) using
valence bond theory?
Note that this time we must start with the orbital diagram for P
3s2
3p3
We easily see that there is no possible way that we can form 5 bonds around the P atom
presently. Therefore, we must use the energetically accessible 3d orbitals.
What happens when we promote the electron?
3s1
3p3
3d1
We mix (hybridise) the s orbital, three 3p orbitals, and a 3d orbital to form five new sp3d
hybrid atomic orbitals.
sp3d hybrid orbitals
unhybridised d orbitals
5 sp3d orbitals these orbitals overlap with the 3p orbitals in Cl to form the 5 bonds
with the required VSEPR geometry trigonal bipyramid.
11
Bond overlaps
type
[sp d (P ) – 3pz (Cl) ] x 5
3
sp3d2 Hybridisation
Example - SF6
6 S-F single bonds (octahedral geometry)!
Note that this time we must start with the orbital diagram for S
3s2
3p4
We easily see that there is no possible way that we can form 6 bonds around the S atom
presently. Therefore, we must again use the energetically accessible 3d orbitals.
What happens when we promote the electron?
3s1
3p3
3d2
We mix (hybridise) the s orbital, three 3p orbitals, and two 3d orbitals to form six new
sp3d2 hybrid atomic orbitals.
sp3d2 hybrid orbitals
unhybridised d orbitals
6 sp3d2 orbitals these orbitals overlap with the 2pz orbitals in Cl to form the 6 bonds
with the required VSEPR geometry octahedral.
Bond overlaps
[sp3d2 (S ) – 2pz (F) ] x 6
type
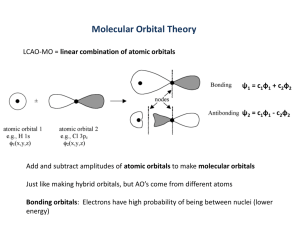
M.O. Theory
Valence bond and the concept of the hybridisation of atomic orbitals does not account for a
number of fundamental observations of chemistry.
Best one O2 - paramagnetic even though the Lewis Structure suggests all oxygen electrons
are paired (paramagnetic). To reconcile these and other differences, we turn to molecular
orbital theory (MO theory). In MO theory, covalent bonding is described in terms of
12
molecular orbitals, i.e., the combination of atomic orbitals that results in an orbital
associated with the whole molecule.
NOTE - as with valance bond theory (hybridisation)
2 AO’s 2 MO’s
E.g., the hydrogen molecule (H2)
We have 2 ls (H) orbitals that interact to form 2 MO’s (a bonding and an anti-bonding MO)
Bonding MO - lower energy and greater stability than the AO’s from which it was formed.
Anti-bonding MO - higher energy and less stability than the atomic orbitals from which it
was formed.
Recall the wave properties of electrons.
constructive interference the two e- waves interact favourably loosely analogous to a
build-up of e- density between the two atomic centres.
destructive interference unfavourable interaction of e- waves analogous to the
decrease of e- density between two atomic centres.
bonding = C1 ls (H 1) + C2ls (H 2)
anti = C1 ls (H 1) - C2ls (H 2)
Bonding Orbital a centro-symmetric orbital (i.e. symmetric about the line of
symmetry of the bonding atoms).
Anti-bonding orbital a node (0 electron density) between the two nuclei.
Bonding orbital for H2, contains both paired e-‘s. i.e., e- density is high about the
nucleus.
13
1s
1s
1s
Energy
1s
The situation for two 2s orbitals is the same! The situation for two 3s orbital is the same.
Let’s look at the following series of molecules
H, H2, He+
Define bond order = ½ {bonding - anti-bonding e-‘s}. Note higher bond order greater
bond stability.
Bond Order
H
H2
He+
He2
# of e- = 1
# of e- = 2
# of e- = 3
# of e- = 4
ls1
1s2
ls2 ls*1
ls2 ls2
The Molecular orbitals for the second row diatomics.
The 2s orbitals.
Bond order =
Bond order =
Bond order =
Bond order =
½ (observed)
1 (stable)
½ (observed)
0 (never observed)
14
2s
2s
2s
Energy
2s
What about the molecular orbitals formed from the 2p atomic orbitals?
2p
2p
2p
2p
Energy
2p
2p
Look at the energies of the energies of the molecular orbitals.
15
1s < 1s* < 2s < 2s* < 2py = 2pz < 2px < *2py = *2pz < *2px
The electron configurations for the second row diatomic molecules.
Li2 number of electrons = 6
Electron configuration = ls2 ls2 2s2
Bond order = ½ (4-2) = 1
Li2 is predicted to be stable and diamagnetic (observed)
Be2 number of electrons = 8
Electron configuration = ls2 ls2 2s2 2s2
Bond order = ½ (4-4) = 0
Be2 is predicted to be unstable (note diatomic beryllium has never been observed)
B2
number of electrons = 10
Electron configuration = ls2 ls2 2s2 2s2 2py1 2pz1
Bond order = ½ (6-4) = 1
B2 is predicted to be stable (single bond) and paramagnetic.
C2
number of electrons = 12
Electron configuration = ls2 ls2 2s2 2s2 2py2 2pz2
Bond order = ½ (8-4) = 2
C2 is predicted to be stable (double bond) and diamagnetic.
N2
number of electrons = 14
Electron configuration = ls2 ls2 2s2 2s2 2py2 2pz22px2
Bond order = ½ (10-4) = 3
N2 is predicted to be stable (triple bond) and diamagnetic.
O2
number of electrons = 16
Electron configuration = ls2 ls2 2s2 2s2 2py2 2pz22px2 2py1 pz1
Bond order = ½ (10-6) = 2
O2 is predicted to be stable (double bond) and paramagnetic.
F2
number of electrons = 18
Electron configuration = ls2 ls2 2s2 2s2 2py2 2pz22px2 2py2 pz2
Bond order = ½ (10-8) = 1
F2 is predicted to be stable (single bond) and diamagnetic.
Ne2 number of electrons = 20
16
Electron configuration = ls
pz22px2
Bond order = ½ (10-10) = 0
Ne2 is predicted to be unstable. Ne is a noble gas (unreactive) and doesn’t want to
combine with other elements to form compounds.
2
ls2 2s2
2s2
2py2
2pz22px2
2py2
17
Delocalised MO’s
So far, we have only discussed localised bonding, i.e., the electrons forming the bonds are
totally associated with the two atoms forming the bond. There are bonding situations where
the electrons are not associated with any one particular atom in the molecules. In such cases,
we refer to the electrons as being delocalised. This is especially true of molecules where
resonance structures can be written.
e.g. benzene, C6H6, resonance structures
resonance hybrid
The C-C bonds are formed from the sp2 hybrid orbitals. The unhybridised 2pz orbital on
C overlaps with another 2pz orbital on the adjacent C atom. This leads to the formation of
three bonds. This overlap, however, extends over the whole molecule (i.e. the bonds
are delocalised). This is in agreement with experiment where all the carbon-carbon bond
lengths and bond orders are the same.
NOTE delocalised bonds (bonds extending over a number of atoms in a molecule) are
generally more stable (less reactive) than molecules whose bonds extend over only 2
atoms.
As these bonds are spread out over the entire benzene molecule, i.e., they are delocalised,
the electrons are free to move around the benzene ring.
We also observe delocalisation in the carbonate anion and the nitrate ion two other species
where we were able to draw resonance structures).