P_-_Hemostasis

Hemostasis (Prohaska)
Zymogen activation is the process that initiates clot formation & clot lysis. It also makes bradykinin, a
vasodilator. (This mechanism is similar to what is seen in the activation of pancreatic digestive enzymes from pancreas gut lumen (STORAGE ACTIVE) & complement cascade)
Hemostasis = stopping bleeding, involves vasoconstriction (of BV), platelet aggregation, and blood clot (fibrin) formation.
Platelets (thrombocytes) are disc-shaped cells, no nuclei, do have mito. Come from megakaryocytes in BM.
10 day life span. JOB = forming mechanical plugs at site of vessel injury & to secrete regulators of clotting
process & vascular repair. The surface binds tightly to collagen (exposed by damaged BVs) via glycoprotein receptors (GpIa). GpIb is another platelet membrane protein that binds tightly to vWF (Von Willebrand factor). Have granules and canalicular system (allowing for larger surface area on membrane). Granules:
electron dense granule = Ca, ADP, ATP & serotonin; α-granule = heparin antagonist, platelet-derived GF, βthromboglobulin, fibrinogen, VWF & other clotting factors; lysosomal granule = hydrolytic enzymes. The cytoskeleton changes shape in response to ADP & thrombin (discs spheres). Platelet has microfilaments of lots of actin & myosin, when injured the Ca release causes contraction changes in cytoskeletal architecture.
Platelet adhesion: o Adhesion = platelet-subendothelial interaction after BV injury o VWF & collagen exposed after BV injury o GPIa + collagen = change in platelet shape to sphere that is able to extrude long pseudopods promoting further platelet interactions o GPIb + collagen = exposure in membrane of
GPIIb/IIIa which are binding sites to fibrinogen and VWF
VWF – made in endothelial cells & megakaryocytes, also binds to Factor VIII in circulation to protect degradation – accelerates clotting. Migrates to surface upon damage.
Platelets help hemostasis by: (SEROTONIN & ADP imp!) o Vasoconstriction – caused by serotonin (released from granules) & thromboxane A2 (derivative of arachidonic acid in the platelet phospholipid membrane)
Prostacyclin is made by vascular endothelial cells to inhibit platelet aggregation. It is a vasodilator.
Works to balance out the behavior of TXA2. Also a derivative of arachidonic acid w/ PGH₂ as an intermediate. o Formation of platelet plug (primary hemostasis) – ADP &
TXA2 stimulate platelet aggregation. Surface lipoproteins of platelets bind clotting factors and activate. Surface GPIIb/GPIIIa binds fibrinogen. o Clot formation – forms around platelets (need membrane phospholipids) by using initial clot factors from granules o Aspirin therapy – will acetylate cyclo-oxygenase, irreversibly deactivating is. COX is the enzyme in the first step of derivatizing arachidonic acid TXA2. Other NSAIDs are reversible inactivators. Isn’t a problem w/ prostacyclin b/c they aren’t made in the portal blood (which is where aspirin takes its
effect on platelets). COX-2 inhibitors can have bad SE on CV function. Long term use of COX-2 inhibitors might not be good b/c it can tilt towards thrombosis over time.
Vascular endothelial cells involved: o Prostacyclin (PGI₂) & NO (from arginine) – these both oppose platelet aggregation & vasoconstriction o Thrombomodulin is a surface protein that binds thrombin. It activates Protein C (& Protein S). o VBF – von Willebrand factor.
CLOTTING:
Overview of clotting see fig 45.3 pg. 860
Fibrinogen Fibrin clot. o Fibrinogen has 3 globular units held together by 2 rods formed by pairs of 3 different polypeptide chains. Central globular region is held by disulfide linkages = disulfide knot. The α and β chains are acidic – have lots of negative charge, so center of fibrinogen molecule has negative charge as floating through blood. o Thrombin (a serine endopeptidase) removes the acidic α and β chains giving it a positive charge in the middle region and producing FIBRIN. o Fibrin can now form a 3D lattice by staggering the molecules. Ca needed for this step & catalyzed by Factor XIIIa = fibrin-stabilizing factor (13). [Factor 13 – only one that isn’t a serine protease].
Fibrin clot is strengthened by cross-linking of interchain glutamine
& lysine. An amide (peptide) bond is formed.
Thrombin formation o Prothrombin has
-carboxyglutamic acid (GLA) residues. GLA residues are important for binding Ca²⁺ and are made posttranslationally in a rxn requiring Vit K.
Vit K is made in liver (hepatocytes) Vit
KH₂ which is a cofactor for addition of carboxyl group to glutamate residues to form prothrombin GLA. During this reaction, Vit K Vit K epoxide that needs to be converted back to Vit KH₂ : this occurs by reduction by Vit K epoxide reductase then back to KH₂. o Factors VII, IX, X & Protein C also have GLA residues (so they also need Ca). o Prothrombin Thrombin reaction is a double cleavage reaction that forms an αβ structure held together by a disulfide bridge. (Occurs in liver)
COMPLEX assembly is needed for this reaction: phospholipid micelles (platelets) & calcium /
Factor Xa (serine endopeptidase, Stuart Factor) / Factor Va (permissive role – accelerin)
Factor Va-platelet complex causes prothrombin to have conformational change making it more susceptible to enzymatic cleavage. Then Factor Xa binds allowing for the conversion of Prothrombin Thrombin to occur.
Complex formation increases rate of Thrombin formation a lot! And…allows thrombin formation to be localized to site of vascular injury (where negatively charged phospholipids are) o Jobs of Thrombin in cell include catalysis of: Fibrinogen Fibrin, V Va (accelerating its own biosynthesis), VIII VIIIa (anti-hemophiliac), XI Xia, XIII XIIIa (cross linking)
Activation of Factor X: o Extrinsic pathway – releases tissue factor from subendothelium. This is rapid.
1. Damage to tissue Tissue factor release
2. Tissue factor binds to VIIa (which is produced by active Thrombin & Xa)
--There is always a little VIIa around
3. VIIa converts X Xa (which can then help make thrombin)
Cross talk bxt Extrinsic & Intrinsic occurs =
VIIa can also activate IX, which will then activate X. o Intrinsic pathway – slower. Involves 3 endopeptidases: Factors XIIa, Xia, IXa, a modifier protein (VIII/VWF) , platelet phospholipids & Ca.
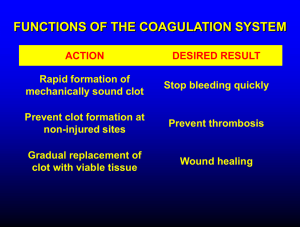
Inhibition of clotting (by plasma inhibitors): o Antithrombin III. Natural inhibitor (a serpin) that inactivates by binding (no hydrolysis) to thrombin IIa mostly. [also to IXa, Xa, XIa, XIIIa]. Reactivity enhanced by heparin – a common anticoagulant that is from the body. o Protein C (& cofactor S). Digests active blood clotting proteins like Va, VIIIa via hydrolysis. Protease. o TFP-1 (Tissue Factor Pathway Inhibitor). Inhibits extrinsic pathway by binding to both Xa & VIIa o Thrombomodulin. On surface of endothelium, binds thrombin which limits clotting ability by activating Protein C.
Fibrinolysis is turned on so once the platelet plug is established it doesn’t continue to aggregate platelets. o Plasmin (which is from plasminogen) is a serum protease that will degrade fibrin. Plasminogen likes to grab on to the surface of a clot even though it can be converted to plasmin in the liquid phase of blood; this allows local action, protection from blood serpins and quicker activation to plasmin. o Plasminogen activating factors facilitate the switch to plasmin
t-PA (tissue plasminogen activator) is made by vascular endothelial cells, high affinity for fibrin o α-2 antiplasmin only works when this is occurring in free blood
Vasodilation is also imp regulator following clot formation.
Vitamin K:
Vit K1 (phylloquinone) – from plant foods & intestinal synthesis. (This is why long term Abx tx can be bad
can cause Vit K deficiency from over killing of ‘good’ bacteria)
Vit K2 (menaquinone) – from animal tissues, converted in liver
Functions: o Is a coenzyme – goes through reduced and oxidized forms that need continual recycling o Makes Ca binding proteins
osteocalcin or bone GLA protein (mobilizes Ca from bone)
Nephrocalcin (Ca resoprtion?) o CLOTTING! Factors II, VII, IX, X, protein C, protein S
We give it to NBs b/c they don’t have a store built up yet
Deficiency: prolonged blood clotting, bruise easily
Antagonists of Vit K: o Dicoumarol – similar to Warfarin o Warfarin (Coumadin) – commonly used rodenticide (eek, kills rats in barns) o Prevents reduction of epoxide o TO prevent clots!
Practice Qs:
Q: Patients who make autoantibodies against platelets are discouraged from taking Aspirin to relieve pain because??
A: Aspirin interferes with platelet function
Q: Vit K antagonists are taken in cardiac patients to:
A: Attenuate clotting (weaken)
1. The edema observed in patients with non–calorie protein malnutrition is due to which of the following?
(A) Loss of muscle mass
(B) Ingestion of excess carbohydrates
(C) Increased fluid uptake
(D) Reduced protein synthesis in the liver
(E) Increased production of ketone bodies
2. A recent surgery patient receiving warfarin therapy was found to be bleeding internally. The clotting process is impaired in this patient primarily because of which of the following?
(A) Inability of the liver to synthesize clotting factors
(B) Specific inhibition of Factor XIII activation
(C) Inability to form clotting factor complexes on membranes
(D) Reduction of plasma calcium levels
(E) Enhancement of protein C activity
3. An inactivating mutation in which of the following proenzymes would be expected to lead to thrombosis, uncontrolled blood clotting?
(A) Factor XIII
(B) Prothrombin
(C) Protein C
(D) Factor VIII
(E) Tissue factor
4. Classical hemophilia A results in an inability to directly activate which of the following factors?
(A) Factor II
(B) Factor IX
(C) Factor X
(D) Protein S
(E) Protein C
5. Hemophilia B results in an inability to directly activate which of the following factors?
(A) Factor II
(B) Factor IX
(C) Factor X
(D) Protein S
(E) Protein C
1. The answer is D. Under conditions of reduced protein ingestion, essential amino acids are scarce and the liver reduces protein synthesis, including circulating plasma proteins. The reduction of protein in the plasma results in a lower osmotic pressure, so excess fluid in the extravascular spaces cannot return to the blood, and remains outside of the circulation, collecting in tissues.
2. The answer is C. Warfarin inhibits the reduction of vitamin K epoxide, so active vitamin K levels decrease. The reduction in active vitamin K levels reduces the γ-carboxylation of clotting factors. In the absence of γ-carboxylation the clotting factors cannot bind to calcium to form membrane-associated complexes with other clotting factors. Warfarin has no effect on the liver's ability to synthesize the clotting factor (the synthesized factor is not modified), nor does warfarin specifically inhibit the activation of factor XIII. The inhibition is more global than just attacking one step in the coagulation cascade. Plasma calcium levels are not altered by warfarin, and protein C activity is actually decreased in the presence of warfarin, because protein C is one of the proteins that is γ-carboxylated in a vitamin K–dependent reaction.
3. The answer is C. Activated protein C turns off the clotting cascade; in the absence of protein C, regulation of clotting is impaired, and clots can develop when not required. Mutations in any of the other answers listed would lead to excessive bleeding, as an essential component of the clotting cascade would be inactivated.
4. The answer is C. Classical hemophilia is absence of Factor VIII, which is a necessary cofactor for the activation of Factor X by
Factor IXa. Factor II is directly activated by Factor Xa, and Factor IX is directly activated by Factor XIa. Proteins C and S are directly activated by thrombin, Factor IIa.
5. The answer is B. Hemophilia B is an inactivating mutation in Factor IX, such that Factor IXa cannot be formed. Factor XIa is formed, but its substrate, Factor IX, is defective, and a nonactive protein results.