Supplementary Information (docx 34K)
advertisement
")
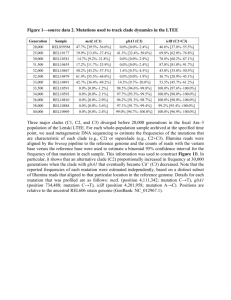
1 Supplementary Material 2 3 The pedigrees (PGs) 1, 2, 3, 4, 12, 13, 15 have been already described. 1-4 4 The characteristics of each new PG and the significance of RUNX1 mutation are described below. 5 Familial trees are presented in Supplementary Figure 1A, the number of patients and PGs are presented 6 in Table 1. 7 * PG5, mutation p.P218S: Two identical twins (patients 10,11) have been diagnosed FPD/AML when 8 they were 5 years old with a thrombocytopenia at 30-40 G/l, the other members of the family couldn’t be 9 studied (refusal). 10 * PG6, mutation p.G108V: The proband (III-1, patient 12) developed a T-ALL at the age of 14 years old. 11 His uncle and his grand-father had thrombocytopenia. The G108V mutation is predicted as deleterious 12 according to prediction software. 5 13 * PG7, mutation p.D305TfsX262: The proband (II-4, patient 13) developed AML at 37 years. She had a 14 medical history of thrombocytopenia with metrorrhagia. Her daughter and her mother have both 15 thrombocytopenia, her three sisters have a normal platelet count. The frameshift RUNX1 mutation occurs 16 in the transactivation domain. 17 * PG8, mutation p.H377PfsX191: The patient (patient 14) had thrombocytopenia discovered at the age of 18 3 years, the other members of the family couldn’t be studied (refusal). She developed AML when she was 19 12 years old and died from infectious complications. The frameshift RUNX1 mutation occurs in the 20 transactivation domain. 21 * PG9, mutation p.G108V: The proband (II-1, patient 15) is thrombocytopenic. Her father died from 22 AML and her sister is thrombocytopenic too. The mutation G108V is predicted as deleterious according 23 to prediction software. 5 24 * PG10, mutation p.G143RfsX43: The proband (I-1, patient 16) developed AML at 36 years. His 25 daughter and his son are both thrombocytopenic with the same RUNX1 mutation. He underwent an 1 26 allograft from his HLA-identical brother, who is not thrombocytopenic and not mutated for RUNX1. The 27 frameshift mutation is in the DNA binding domain of RUNX1. 28 * PG11, mutation p.T169R: The proband (I-1, patient 17), his two daughters (II-2,3, patients 18,19) and 29 his son (II-1, patient 20) are all thrombocytopenic and mutated for RUNX1, with bleeding history. This 30 mutation is predicted as deleterious according to prediction software. 5 31 * PG14, mutation p.T121HfsX9: The proband (I-2, patient 23) has thrombocytopenia. His son (II-1, 32 patient 24) has thrombocytopenia with bleeding history. He developed AML at the age of 6. They are 33 both mutated for RUNX1, with a frameshift mutation occurring in the DNA binding domain. 34 35 References 36 37 38 1. Preudhomme C, Renneville A, Bourdon V, Philippe N, Roche-Lestienne C, Boissel N, et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 2009 May 28; 113(22): 5583-5587. 2. Beri-Dexheimer M, Latger-Cannard V, Philippe C, Bonnet C, Chambon P, Roth V, et al. Clinical phenotype of germline RUNX1 haploinsufficiency: from point mutations to large genomic deletions. European journal of human genetics : EJHG 2008 Aug; 16(8): 1014-1018. 3. Bluteau D, Gilles L, Hilpert M, Antony-Debre I, James C, Debili N, et al. Down-regulation of the RUNX1-target gene NR4A3 contributes to hematopoiesis deregulation in familial platelet disorder/acute myelogenous leukemia. Blood 2011 Dec 8; 118(24): 6310-6320. 4. Heller PG, Glembotsky AC, Gandhi MJ, Cummings CL, Pirola CJ, Marta RF, et al. Low Mpl receptor expression in a pedigree with familial platelet disorder with predisposition to acute myelogenous leukemia and a novel AML1 mutation. Blood 2005 Jun 15; 105(12): 4664-4670. 5. Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS computational biology 2014 Jan; 10(1): e1003440. 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 2