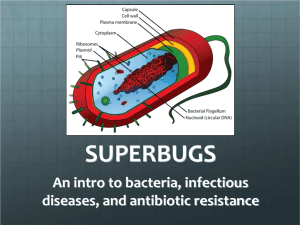
Learning Objectives for Disease and Defense
advertisement

Learning Objectives for Disease and Defense Week 1 Absorption and Distribution 1. Identify the factors that determine a given drug’s ability to cross biological membranes. a. Molecular size: smaller MW drugs will be absorbed more readily, size also affected by the drug binding to plasma protein (increases size, decreasing absorption) b. Lipid solubility: increased lipid solubility leads to increased absorption (drug can easily cross lipid bilayer of membranes), estimated by oil:water partition coefficient c. Degree of ionization: affected by pH, will influence lipid solubility (more unionized=more lipid soluble=increased absorption), requires H-H equation d. Concentration gradient: high concentration created at site of drug administration, drug will move from [high] to [low]. 2. Describe the mechanisms by which drugs cross biological membranes (diffusion, transport, etc.). a. Passive diffusion i. MOST IMPORTANT ROUTE, driven by concentration gradient ii. Aqueous diffusion/filtration (drug flows through aqueous channel): limited capacity, channel size varies (generally for drugs of MW less than 100-200) iii. Lipid diffusion (drugs pass via hydrophobic bonding with membrane lipids): favored if drug has high lipid:water partition coefficient, often pH dependent, unionized moiety crosses membrane down concentration gradient, most important mechanism for drugs with MW of 500-800. b. Carrier mediated diffusion i. Facilitated diffusion: driven by concentration gradient (no energy required) ii. Active transport: energy dependent, selective, saturable, unidirection, for durgs which resemble endogenous compound. Many cells also contain less selective membrane transporters that are specialized in expelling foreign molecules (ie: P-glycoprotein). Drugs are inhibitors of these transporters can be involved in certain drug-drug interactions via alteration of substrate drug levels in tissues. c. Endocytosis i. Of minor importance to drug passage, pinocytosis or phagocytosis 1 3. Summarize the therapeutic advantages and disadvantages of the various routes of drug administration, especially with regards to bioavailability and rate of onset of effect. ROUTE BIOAVAILABI RATE OTHER FACTORS LITY Enteral (GI Oral 0-100%: Slow (15-30 Most common, drug absorption occurs via passive tract) depends on min for diffusion (favors lipophilic/nonionized drugs), rate of survival in GI immediate absorption of drug from intestine ˃stomach (because of environment, release) large SA of intestine), increased GI motility and empty ability to stomach=increased absorption, duration: min-hrs cross GI Slower Enteric drug coat: protects stomach from irritation and membrane, (hours) for protects drugs from low stomach pH efficiency of enteric/sust Controlled-release prep: rate of absorption is slowed by drug ained slowing rate of product dissolution (allows for fewers metabolism release) administrations, increased compliance, overnight by gut wall or therapy, elimination of peaks/troughts, BUT greater st liver (1 interpatient variability and formulation could fail giving pass) pt entire dose “dose dumping” Enteral (GI Rectal Variable, but Not rapid Useful with oral route is unavailable (vomiting, tract) generally ˃ unconscious, post-GI surg, uncooperative pt), 50% of oral dose will BYPASS liver (first pass metabolism is ˂ for oral, absorption is irregular/incomplete Parenteral Sublingual Generally Within Absorbed from mouthSuperior vena cava (protects (Outside GI) Buccal high minutes (5- drugs from hepatic 1st metabolism + faster onset), useful 10 min) for drugs that are lipid soluble and relatively potent (˂1mg dose) as there is smaller SA for absorption relative to GI tract Parenteral Intravenous 100% MOST Rapid Most direct route, circumvent all factors related to (Sec-min) membrane passage/absorption, accurate and fast drug delivery, used for drugs with narrow therapeutic window, requires aseptic technique, most haxardous route b/c easy to reach irreversible toxic levels quickly, duration= t1/2-dep Parenteral Intramuscular Approaches Aqueous Absorption/onset effected by blood flow/muscle activity 2 100% soln-rapid (5-10 min), slower in depot form 5-10 min, Slower, constant rate for depot (hours) Rapid (˂5 min for gaseous)Ne gligible Parenteral Subcutaneous Approaches 100% Parenteral Inhalation Gaseous: Gas/volatile liquids OR Suspension: Aerosol or microparticles Variable100% a. about 100% b. variable Parenteral Transdermal at site of injection, depot form in oil or suspended= slower/sustained absorption, used if drug is too irritating for subQ, absorption may be erratic/incomplete with solubility is limited, disadvantage: pain, tissue necrosis (if high pH), microbial contamination Only for non-irritating drugs, volume of dose is limiting, period of drug absorption can be altered via particle size, protein complexation, pH (insulin), addition of vasoconstrictor (local anesthetics), pellet implantation (contraceptives) Gas: used for rapid onset of systemic drug effects (nicotine, crack, general anesthetics), rapid rate of absorption due to large SA and high blood flow in pulmonary tissue Particle: applied at site of action in lung, increases local topical effects (reduces systemic effects), exampleasthma, depends on particle size < 0.5 M: exhaled from lungs no effect 1-5 M: deposited in small airways therapeutic > 10 M: deposited in oropharynx side effects Irritant drug may induce bronchospasms Slow (hours) Topical Apply patch to skin for tx of systemic conditions, prolonged drug levels attained, 1st pass metabolism is avoided, drug must be potent (dose ˂2 mg), must permeate skin w/o irritation, examples: estrogens, testosterone, fentanyl, nicotine, nitroglycerin Localized application via skin/mucous membrane (vaginal, nasal, eye) for tx of local conditions, minimal systemic absorption, in children potential for 3-fold greater system availability that in adult (body SA: weight is greater) 4. Explain the influence of pH on the ionization of weak acid / weak base drugs. a. Most drugs are either weak acids or weak bases, therefore they are present in biological fluids are ionized or non-ionized species. b. Non-ionized forms are more readily absorbed. Ionized forms DO NOT cross lipid membranes. c. Weak Acids: i. HA (or R-COOH) ↔ H+ + A- (R-COO-) ii. R-COOH is the protonated/non-ionized form of the acid and can cross biological membranes (in acidic environment) iii. R-COO- is the un-protonated form of the acid and will be “ion-trapped” d. Weak Bases: i. BH+ (R-NH3+) ↔ H+ + B (R-NH2) ii. R-NH3+ is the protonated form of the base and will be “ion-trapped” 3 iii. R-NH2 is the un-protonated/non-ionized form of the base and will cross biological membranes (in basic environment) 5. Be able to use the Henderson-Hasselbach equation to qualitatively predict the ratio of ionized to unionized species of a weak acid or weak base drug in various body compartments. a. HH equation: determines the extent of ionization of an acid or a base (dependent on the strength of the acid or base (pka) and the pH of the body fluid). Allows quantitation of the fraction of the total amount of drug that is ionized or unionized and allows predictions of a pH that at which the majority of the drug will be non-ionized and thus will be absorbed. b. pH-pKa= log (non-protonated: A- or B)/ (protonated: HA or BH+) i. pH= pH of the biological compartment the drug is in ii. pKa= pH of a solution at which concentrations of the protonated and unprotonated forms of the drug are equal. c. If pH is lower than pKa (lots of protons): protonated form of weak acid (unionized-lipophilic) or weak base (ionized) will predominate d. If pH is higher than pKa (fewer protons): unprotonated form of weak acid (ionized) or weak base (unionized) will predominate. e. “Ion Trapping”: lipid barriers may separate two aqueous solutions with different pH’s. Only non-ionized drugs can diffuse through membrane and this form of the drug will equilibrate and be the same on both sides of the membrane. At equilibrium, un-ionized concentration of drug is the same on both sides of the membrane, but total concentration of drug is greater on the side where ionization is greater. i. Acidic drugs will be trapped in BASIC solutions. Basic drugs will be trapped in ACIDIC solutions. They are trapped where they are predominantly ionized. ii. Clinical significance: altering urinary pH to ion trap weak acids or bases and hasten renal excretion (in aspirin overdose situations), greater potential to concentrate basic drugs in more acidic breast milk 6. Explain the therapeutic consequences of anatomic “barriers” to distribution and selective accumulation of drugs. a. Tissues with tight junctions between cells (GI mucosa, BBB, placenta, renal tubules), require that drugs pass through lipid membranes to or from this compartment and into or out of the blood. Drugs that 4 can’t pass through membranes (large size, protein bound, highly charged, high water solubility) will be UNABLE to move between these compartments and blood. i. GI mucosa: negligible absorption of drug into blood in administered orally ii. BBB/placenta: limited distribution of drug from blood into brain or into fetal circulation, due to structural differences between brain and non-brain capillaries i. Renal tubules: following filtration at glomerulus (note that large or protein bound drugs are NOT filtered), reduced reabsorption of drug back into blood, thus enhancing excretion via urine. ii. Selective accumulation of certain drugs may occurs in specific tissues and can be harmful or beneficial (kidney, eye, lung, bone, ear) 7. Describe how drug binding to plasma proteins can effect drug distribution and elimination as well as be a potential source of drug-drug interactions. a. Will influence distribution as only FREE DRUG is diffusible. b. Acidic dugs bind to albumin and basic drugs bind to alpha-1 acid glycoprotein c. As drug binding to protein increases: i. Decreased concentration of free drug (can limit fetal exposure to drug) ii. Decreased metabolic degradation and rate of excretion (will decrease elimination rate and increase half-life), acis as circulating drug reservoir that can prolong drug action iii. Decreased volume of distribution by enhancing apparent solubility in blood (because only free drug can get out) iv. Decreased ability to enter CNS across BBB (because only free drug enter brain more readily) d. Mediates protein binding/displacement drug-drug interactions i. Displacement of 1st drug from protein binding site by 2nd drug results in increased levels of unbound 1st drug, but levels of total drug are unchanged because administration is unchanged. e. Unlikely to be of clinical significance unless the displaced drug has narrow therapeutic index, displacing drug is started in high doses, Vd of displaced drug is small, or response to drug occurs more rapidly than redistribution. 5 8. Explain the derivation and clinical relevance of the following pharmacokinetic parameters. Describe their use in designing dosage regimens: a. Bioavailability (F): Adjustment of dose for oral vs parenteral administration. i. Bioavailability (F or f[%]): fraction of unchanged drug reaching the systemic circulation, determined by comparing AUC following single dose of drug given by any route to the AUC following single dose by IV route. ii. F = Fraction Bioavailable = AUCORAL / AUCIVA iii. Information on extent of absorption (bioavailability) by ORAL route is available for most drugs. You can use this for dosage adjustments when drug is given by a different route (common to switch from oral to IV). iv. F= 100% for IV, F=0-100% for ORAL, F= about 100% for other system drug action routes b. Volume of distribution (Vd): Converting drug dose to plasma concentration, selecting loading dose, implications of high or low values. i. Vd: size of compartment necessary to account for total amount of drug in the body if it were present throughout body at same concentrations found in plasma. It is the volume of the body fluids into which the drug distributes following administration. Gives indication of the extent to which a drug passes from plasma to extravascular tissues ii. High values of Vd indicate drugs located mostly outside of plasma (increased tissue binding, high lipid solubility) iii. Low values of Vd indicate drugs located mostly inside the plasma or ECF (extensive binding to plasma proteins or large size) iv. Vd varies between patients due to: Body Weight, Fat vs. Lean and Changes in Protein Binding v. Vd allows determination of the necessary single dose of drug (loading dose) to fill the distribution volume with enough drug to achieve desired steady state level (Cp) vi. Vd also allows prescriber to determine effect of any given dose (D) will have on the plasma concentration 6 Examples: a. b. c. d. 1. Loading Dose (LD)= Cp (desired) x Vd 2. Cp= Dose/Vd 3. Vd (L)= DOSE (mg)/concentration of drug in plasma (mg/L) 4. DOSE (mg)= concentration of drug in plasma (mg/L) x Vd (L) 5. Concentration of drug in plasma (mg/L)= DOSE (mg)/Vd (L) Drug A, Dose (mg)= 1000, Cp0 (mg/L)= 333 i. Vd (L)=3, most drug stays in plasma Drug B, Dose (mg)=1000, Cp0 (mg/L)= 66 i. Vd (L)=15 Drug C, Dose (mg)=1000, Cp0 (mg/L)=25 i. Vd(L)= 40 Drug D, Dose (mg)=1000, Cp0 (mg/L)= 2 i. Vd(L)= 500, most drug leaves plasma Metabolism and Excretion 1. Describe the general principles and consequences of drug metabolism. a. Drug metabolism: drugs undergo enzyme-catalyzed chemical structure transformation after administration to the patient (if only terminated only by renal excretion, the duration of action would be prolonged) b. Drug metabolizing enzymes have endogenous substrates and play a role in normal metabolism c. Liver is primary site of drug metabolism, but lungs (30%), intestines (6%), kidney (8%), skin (1%), placenta (5%) and bacteria have enzymes capable of drug metabolism d. Oxidation is most common pathway, but other types of chemical transofmration can occur. Many transformation are catalyzed by membrane-bound enzymes of the SER called CYP450 e. Lipid-soluble compounds are generally converted to more H20-soluble (more polar) compounds that are more readily excreted f. Generally, metabolism is detoxifying process (active drug to inactive or less active compound), but can also metabolize active drug into MORE active compound (ie: codeine morphine), metabolize inactive compound into active ingredient, metabolize to toxic metabolite 2. Describe the general characteristics of Phase I (oxidation [CYP450 and non-CYPP450 (non-microsomal)], reduction, hydrolysis) and Phase II reactions (conjugations: glucuronide, sulfate, glycine, glutathione) as related to: Qualitative and quantitative role in drug metabolism Classifications of reactions [ex., O-dealkylation is phase I oxidation (P450)] PHASE I: inserts or unmasks a functional group on the drug that renders molecule more water-soluble and the molecule can then undergo conjugation in Phase II rxn. 7 PHASE II: endogenous substrate combines with pre-existing or metabolically inserted functional group on the drug forming a highly polar (water soluble) conjugate that is Reactions Enzymes involved Genetic Polymorphisms General developmental patterns of activity and age-related changes in activity Inhibitory/Inducibility Relative ease of saturability at high drug substrate levels 1. Oxidation—P450 dependent or P450 independent (most common) 2. Reductions (azo, nitro, carbonyl reductions) 3. Hydrolysis 1. CYP450 (includes NADPH, flavoprotein NADPH-cytochrome P450 reductase, and O2) or non-CYP450 2. Reductase 3. Esterases or amidases YES Examples: Amplichip test available to detect polymorphisms in CYP2D6 / 2C19 Lab tests available to detect genetic variation in anticoagulant response. Warfarin metabolizing enzyme: CYP2C9. Warfarin target enzyme: vitamin K reductase [VKORC1] YES (decreases with age in 1/3 of pts) YES/YES Minimal excreted via the urine. Conjugations: 1. Glucuronidation 2. N-acetylation 3. Glutathione conjugation 4. Sulfate conjugation 1. Transferases (ie: glucuronyl transferases, N-acetyltransferases) Yes (less) Yes (especially UGT) YES (less) Substantial Limited supply of reactants renders Phase II reactions more easily saturable (become zero order elimination kinetics) than phase I reaction 3. Explain the therapeutic consequences of induction and inhibition of metabolism. List the clinically relevant inhibitors and inducers on page 8 of the drug metabolism notes. a. Induction: increased drug metabolizing activity (increased clearance) via stimulation of the CYP450 system, compound that causes induction is an “inducer”, mechanism is often increased synthesis of enzyme protein accompanied with increase in liver weight, proliferation of SER, increases in NADPH and cytochrome P450. Requires 48-72 hours to see onset of effect (slow compared to inhibition). a. Therapeutic consequences: maximal effects seen in 7-10 days, production of pharmacokinetic tolerance (induction by a drug of its own metabolism), induction by 1 agent may increase clearance of another drug reduced therapeutic effect (via increased elimination) or increased toxicity (via toxic metabolite) b. Inhibition: decrease clearance of drug by inhibiting drug metabolizing activity, phase I enzymes are more prone to inhibition compared to Phase II enzymes. Mechanisms: inhibit enzyme synthesis, inhibitor can act as a substrate competing for the enzyme, inhibitor can be an ihbitor without being a substrate, inhibition results from formation of metabolite that destroys enzyme via covalent bonding or form tight complex with enzymes inhibiting its activity. 8 a. Therapeutic consequences: inhibition of metabolism can occur as soon as sufficient hepatic concentration is reached (within hours), inhibition by 1 agent of the metabolism of another can result in decreased clearance of the inhibited drug increased toxicity. INDUCERS (increased clearance) INHIBITORS (decreased clearance) Phenobarbital [1A2, 2C9, 2C19, 3A4] Cimetidine [2D6, 3A4, 1A2] Phenytoin [2C9, 2C19, 3A4] Erythromycin / Clarithromycin [3A4] Carbamazepine [2C9, 2C19, 3A4] Azole antifungals [3A4] Rifampin [1A2, 2C9, 2C18, 3A4] Fluoxetine (other SSRIs) [2D6,3A4] Ethanol [2E1] Grapefruit juice [3A4] St. John’s Wort [3A4] HIV protease inhibitors [3A4] Tobacco smoke (not nicotine) [1A2] Omeprazole [2C19] 4. Describe the general characteristics of drug excretion by the kidney (filtration, secretion, reabsorption and the influence of pH and protein-binding on these processes). A. Excretion: loss of chemically UNCHANGED drug from the body. Kidneys are MOST important organ for excretion (Especially for water-soluble and non-volatile compounds). B. Filtration: glomerular, rate of 120ml/min, all drugs smaller than albumin (MW=69000) will be filtered, only free drug is filtered (NOT protein bound), renal excretion affected by renal blood flow and renal function, drugs cleared by this route have t1/2= 1-4 hours (but high protein binding can extend half-life) C. Secretion: active tubule secretion, drugs transported from blood to urine, rate of 120-600 ml/min, occurs with drugs that are stronger acids and bases in proximal tubule via secretory mechanism that are saturable (examples of drug substrates for transporter= acids: penicillins, salicylate, diuretics, bases: morphine, catecholamines, histamine), plasma protein binding (reversible) does NOT appreciably affect rate of secretion (t1/2= 1-2 hours), poorly developed process in neonates (therefore prolonged half- lives). D. Tubular Reabsorption: drugs that are lipid-soluble and uncharged WOULD be cleared at rate of urine formation (1 ml/min) but the primary function of drug metabolism is to produce more water soluble metabolite that is less likely to be reabsorbed. In order to be reabsorbed, drugs must pass through membranes. i. Passive diffusion occurs with lipid soluble molecules in proximal and distal tubules, as water is reabsorbed, lumen to blood back-diffusion is favored as drug is concentrated in luminal fluid. a. Diffusion of weak acid/bases dependent upon urine pH (non-ionized form only will diffuse across membrane), can change urinary pH with NH4Cl (acidify) or NaHCO3 (alkalinize) ii. Active reabsorption is particularly important for endogenous compounds (glucose, aa’s), most drugs REDUCE this active transport. 5. Describe the therapeutic implications of enterohepatic recirculation of drugs. a. Enterohepatic Recycling: Drug metabolites in liver (usually conjugates that increase MW to ˃300) are secreted into bile, stored in gallbladder, delivered to intestine via bile duct, hydrolyzed by bacterial enzymes back to the parent drug (more lipid soluble) and undergo reabsorption from the gut 9 b. Reduces the elimination of the drug and prolongs its half-life and duration of action in the body. c. Some drugs have “reservoir” of recirculating drug that accounts for 20% of total drug present in the body. d. Antibiotics that reduce gut glora can decrease enteroheptaic recycling and decrease plasma drug levels and is a potential mechanism for drug-drug interaction 6. Describe the factors influencing drug passage from plasma to breast milk. a. Most drugs do cross (unchanged) into breast milk, but at LOW levels. Resulting infant plasma levels is substantially lower than therapeutic levels. To prevent infant exposure: i. Desynchronize breastfeeding and peak milk/drug concentration 1. Breastfeed at end of dosing interval or administer after nursing 2. Administer a dose prior to infant’s longest sleep time 3. Fat (and thus drug) content of milk: increases during feeding period ii. Choose Medication for breastfeeding mother carefully 1. Select drugs with little-no passage into breast milk 2. Drugs with rapid clearance 3. Milk is MORE acidic than plasma (5.6 vs 7.4 pH) and has tendency to accumulate basic compounds by ion trapping 4. Lipid soluble compounds= increased milk concentration 5. High protein binding= decreased milk concentration 6. Never use drugs contraindicated by American Academy of PEDS 7. Drugs that can affect milk production/secretion/ejection (through prolactin, oxytocin) Pharmacokinetics of Elimination 1. Explain the derivation and clinical relevance of the following pharmacokinetic parameters and be able to use them in designing dosage regimens and predicting changes in drug plasma levels and drug response: a. Clearance (CL): Selecting maintenance dose, dosage adjustments necessitated by alteration of kidney or liver function. i. Clearance: the volume of plasma which is completely cleared of drug in a given period of time by the processes of kidney excretion and drug metabolism (with some contribution from other tissues). 10 b. c. d. e. 1. Clearance = volume of distribution (Vd) x Ke a. Ke: fraction of drug eliminated per unit time ii. Clearance: proportionality constant that makes the average plasma concentration at steady state equal to the rate of administration 1. Maintenance dose/tau= CL x Cp(ss) 2. CL= (MD/tau)/ Cp(ss) Half-life (t1/2): Time to steady state or removal from body, selecting dosage intervals, relation to fluctuations in plasma drug levels between drug doses (difference between Cp max and Cp min). i. The time required to eliminate ½ of the drug amount present in the body 1. Drug with T1/2 of 3 hours, takes 3 hours for drug concentration to go from 1000 mg/ml to 500 mg/ml and another 3 hours from drug to go from 500 mg/ml to 250 mg/ml. ii. Time it takes for drug to be essentially eliminated (4-5 half-lives) iii. Time it takes to reach steady state when drugs administered continuously (4-5 half-lives) iv. Degree of fluctuation between doses= 2x, where x=# of t1/2 in T v. t1/2 = 0.693/ke Elimination rate constant (ke) i. ke is the fraction of drug leaving body per unit time via all elimination processes. ke is best thought of simply as a number or constant that allows us to calculate the amount of drug remaining at any time during the elimination process. First-order: Implications for chronic dosing regimens i. Virtually all drugs eliminated via first-order kinetics which means the rate of elimination (mg/hr) is proportional to the concentration of drug in the plasma (mg/L) 1. If concentration of drug is doubled the rate of elimination is doubled ii. As drug is eliminated from the body, its concentration is CONSTANTLY CHANGING, therefore rate of elimination also changes constantly iii. CONSTANT FRACTION of drug is eliminated per time and this is INDEPENDENT OF THE TOTAL AMOUNT OF DRUG PRESENT iv. Most clinically used drugs are eliminated by first-order kinetics when given in doses within the therapeutic dosage range because the major biological processes responsible for drug elimination, hepatic metabolism and renal excretion, are first-order processes. Zero-order kinetics: Implications for chronic dosing regimens. i. Process in which the rate of elimination of drug from the body is INDEPENDENT of the amount of drug in the body. The amount of drug removed per unit time is constant. ii. Most often occurs due to saturation of hepatic metabolic enzyme systems by drug administration. This enzyme saturation occurs with therapeutic doses for only a FEW drugs (aspirin, phenytoin, EtOH) and with toxic doses for most hepatically eliminated drugs. Drugs eliminated by zero-order kinetics don’t have half-lives, can present dose adjustment challenges especially at the upper end of the therapeutic range of narrow therapeutic index drugs, where a small change in dose can produce large changes in plasma concentrations and subsequent toxicity. 11 12 Principles of Pharmacodynamics 1. Describe the drug-receptor concept and its consequences for pharmacotherapy. a. Receptor: component of the biological system to which a drug binds to bring about a change in the function of the system, specificity of the fit of drug to receptor (recognition) induces conformational change in receptor protein. b. Transduction: conformational change in the receptor leads to the transduction that alters cellular function via effector molecules. Effectors accomplish the biologic effect after being activated by the receptor (they translate the drug-receptor interaction into a change in cellular activity). Examples: ligand-gated ion channels (fast response that changes membrane potential), G-protein-coupled receptors (fast response that produces 2ndary messengers, IP3, cAMP, cGMP), kinase linked or hormone (nuclear) receptors (slow response that changes gene expression/protein synthesis. c. Types of Receptor Molecules: i. Specialized: membrane proteins or ion channels designed to detect chemical signals and initiate a response via signal transduction pathways ii. General: biological molecules with any function including enzymes, lipids or nucleic acids. iii. Proteins: binding site for majority of drugs, great specificity (due to secondary and tertiary structure), example: hormone and neurotransmitter receptors, receptor or voltage gated ion channels, enzymes, transport proteins, structural proteins iv. Nucleic acids and membrane lipids: lower specificity d. Consequences of Drug Receptor Therapy i. Receptor mediate the actions of pharmacologic agonists and antagonists 1. Agonists: bind to and regulate function or receptor macromolecules in the same manner as endogenous ligand promoting that receptor function 2. Antagonist: binding to receptors, but unable to generate characteristic response, effect results from preventing the binding of endogenous agonist and blocking their action, will have NO EFFECT in absence of the agonist for that receptor (extent of effect depends on normal “tone”) ii. Receptors are responsible for selectivity of drug action 1. Size, shape, electrical charge, etc. determine binding affinity of drug to particular receptor relative to many other binding sites on patient iii. Theory allows determination of quantitative relation between dose or concentration of drug and its pharmacologic effects via use of dose-response curve 1. Quantify relationship between drug dose and effect helps to understand the drugreceptor interactions. Knowing potency and therapeutic efficacy is important when choosing a drug. e. 2. Explain the theoretical aspects and therapeutic consequences of the hyperbolic shape of the dose-response curve. a. Drug receptor theory assumes that interaction follows simple mass action relationships, binding is reversible and that response is proportional to receptors [R] occupied by drug [D] b. R+ D ↔ R-D R-D is proportional to response c. Dose response curves generated by giving increasing doses of drug and measuring the specified response to each dose d. Hyperbolic shape of curve confirms the mathematical relationship between dose and response as show by this equation: 13 i. e/Emax = [D]/ EC50 + [D] = [D]/ED50 + [D] e. Therapeutic consequences: i. Curve is relatively linear (straight) at low doses meaning that at low doses the response increases in direct proportion to the dose, this is consistent with receptor theory that states that the great the number of receptors occupied by drug, the greater the response provided. ii. Curve levels off at high drug doses meaning that there is a limit to the increase in response that can be achieved by increasing the drug dose, consistent with theory that says the response by administration of drug is proportional to amount of receptors occupied by drug (HIGH dose=all receptors occupied, no further increase can be achieved and Emax achieved) 3. Describe the advantages of the log dose-response curve versus the dose-response curve. a. Advantages of log dose-response: i. Allows for wide range of doses to be plotted allowing easy comparison of different drugs ii. Dose-response relationship is nearly a straight line over large range of doses (corresponds to therapeutic range) 4. Explain the terminology of log dose-response curves: a. Potency: affinity / Kd / EC50 i. Potency: concentration (EC50) or dose (ED50) required to produce 50% of that drug’s individual maximal effect, depends on affinity (Kd) of receptors for binding the drug and in part of the efficiency of this drug-receptor complex to generate a response and is designated by EC50. Provides information on how much drug (dose) will be required to produce a given effect (more potent, less drug needed). EC50 values used to compare potencies of different drugs. b. Efficacy: power / Emax i. Maximal effect or maximal efficacy (Emax): limit of the dose-response relationship on the response axis (y-axis), indicates the relationship between binding to the receptor and the ability to initiate a response. Most important determinant of drugs clinical utility. Power is often used interchangeably with efficacy to describe the ability to initiate a response. c. Agonist - Partial agonist - Antagonist i. Agonist: drug that activates its receptor upon binding and brings about the characteristic tissue response ii. Partial agonist: occupy the same receptor as full agonist, but bring about less than maximum response even at full dosage levels. These are less efficacious. iii. Full agonist: occupy receptors and bring about a full or maximal response (max response being defined as that produced by the most powerful agonist in that tissue) 14 5. Explain the use of log dose-response curves to compare potency and efficacy of different drugs. a. Figure A: EC50x (half maximal effective concentration of drug x) occurs at a concentration that is 1/10 the half-maximal effective concentration of drug y, EC50y. Therefore, drug x is MORE potent than drug y. b. Figure B: Emax for x is 100%, Emax for y is 50%. Therefore, drug x is more efficacious than drug y. Drug y is a partial agonist. 15 c. Potency: Based on position of curve along the dose [x] axis. Drugs A and B are more potent than Drugs C and D. Drug A is less potent than Drug B (partial agonist) because Drug A has a larger EC50 compared to Drug B. Potency: B ˃A˃C˃D. For therapeutic purposes, the potency of a drug is expressed in dosage units for a particular therapeutic endpoint (not as EC50s) i. 20 mg of lisinopril will lower BP by 10-15 mm Hg, as will 50 mg of captopril ii. 200 mg of ibuprofen will alleviate headache pain, as will 650 mg of aspirin d. Maximal Efficacy: this is the limit of the dose-response relation on the response [y] axis. Drugs A, C and D are full agonists with greater maximal efficacy than drug B, a partial agonist. Efficacy refers to the extent a given clinical effect can be achieved in an intact patient (not action at specific target). Efficacyemax: A=C=D˃B. Efficacies of different drugs are compared even though they act at different receptors or targets: i. Furosemide (inhibits Na+-K+-2Cl- cotransporter) is a more efficacious diuretic than hydrochlorothiazide (inhibits Na+-Cl- cotransporter) ii. Morphine (mu opioid receptor agonist) is a more efficacious analgesic than aspirin (inhibits cyclooxygenase) 6. Distinguish between characteristics of the different types of antagonism (pharmacological [competitive reversible and noncompetitive irreversible], physiological, chemical) and provide examples. a. Antagonism: antagonist is a drug that inhibits the action of an agonist but has no effect in the absence of an agonist, divided into receptor and non-receptor antagonists. b. Pharmacological antagonists (aka: receptor anagonists): bind to the same receptor as the agonist. i. Competitive reversible antagonist: binds reversible to the active site of receptor, but does not stabilize the conformation change required for receptor activation. Antagonist blocks agonist from binding to its receptor and maintains receptor in inactive conformation. EC50 increases (potency decreases) - Emax unchanged (because agonist concentration can be increased to outcompete the antagonist). Example: metoprolol is competitive reversible antagonist of NE in Beta1 receptors in heart that produces a reduction in heart rate. But increase in endogenous NE during exercise will still increased heart rate 16 ii. Noncompetitive irreversible antagonist: binds irreversibly (covalently) or pseudo irreversibly (with very high affinity-slow dissociation) to the active site of the receptor. Removes functional receptors from the system, limiting the number of available receptors that can contribute to the response. Noncompetitive because an irreversibly bound active site cannot be outcompeted even at high concentrations). The curve shifts downwards and the maximal efficacy (Emax) of agonist is reduced. No shift on the x-axis. iii. Noncompetitive Allosteric Antagonist 1. Drug binds to different site on the receptor than agonist drug. They do no compete with agonist for receptor binding, but inhibit receptor from responding to agonist, so curve shifts downward, result in a dose-dependent decrease in maximal efficacy (Emax). High concentrations of agonist cannot active the receptor. May see a shift to the right (decrease in apparent potency) if spare receptors available. 17 c. Non-receptor antagonists: include physiological antagonists that bind to a different receptor and chemical antagonists that bind the agonist molecule directly (do not involve any receptor binding). i. Physiological: activates or blocks a distinct receptor that mediates a physiologic response that is opposite to that of activation of the receptor for agonist. ii. Chemical: does not involve receptor binding, antagonism occurs via inactivation of agonist itself by modifying it or sequestering it so it is no longer capable of binding to and activating the receptor. Examples: EDTA chelator, antacid neutralizes stomach acid, osmotic diuretics. Adverse Drug Reactions (Toxicology) and Poisoning 1. Compare and contrast graded dose-response curves and population dose-response curves and explain the use of population dose-response curves to evaluate drug safety (Therapeutic Index and Standard Safety Margin). a. Dose-response curve: a number of increasing doses of a drug are given to the same subject and the increase in response for each dose is measured (graded in increments) allowing determination of the maximal effect of the drug (Emax). ED50 is the dose that produces 50% of the maximal response possible in an individual. b. Population dose-response curve (quantal): characterize pharmacologic responses that are all-or-nothing events (not graded) in a population of subjects (not an individual), generated by arbitrarily defining 18 some specific therapeutic effect (ie: relief of headache) and then determining the minimum dose to produce this response in each member of the population. Single given dose of drug in an individual test subject will either bring about the response or not (all-or-nothing). Data is plotted as fraction of population that response at each dose of drug vs. the log of dose administered. Quantal curves are not used to determine Emax (like dose-response). ED50 is the dose that initates the response in 50% of the test population. c. Degree of risk evaluated by comparing the quantal dose-response curves for the desirable and toxic effects. Generated in the same way except for that the all-or-nothing effect is a toxic effect (side effect, death) and TD50 is the dose that produces an undesirable side effect in 50% of subjects and LD50 is the lethal dose that causes death in 50% of subjects. i. To compare the dosage necessary for desired effect vs. dose with undesirable effect use: 1. Therapeutic index: compares midpoint in the population (ED50 and LD50) a. TI= LD50/ED50 b. The higher the TI, the safer the drug, clinically used drug are ˃10-20 2. Standard safety margin: looks at the extremes in the population (ED50 and LD1) a. SSM= [(LD1/ED99)-1] x 100 b. More conservative measure that TI, more reliable if pt response to therapy to specific drug varies, takes into account the extremes, SSM can be negative. 2. Describe the general FDA categories for drug use in pregnancy and the implications for drug prescribing. a. Used to classify risk to fetus of using drugs during pregnancy. i. A: controlled studies show no risk, possibility of harm to fetus is remote, KCl ii. B: no evidence of risk in humans (opioids, acetaminophen, ondanstron, thiazide diuretics) iii. C: risk cannot be ruled out (pseudoephedrine, antidepressants) iv. D: positive evidence of human fetal risk, but potential benefits may outweigh the risks (in a lifethreatening situation or serious disease) (oral anticoags, ACE inhibitors, diazepam-lorazepan, alprazolam, paroxetine) v. X: contraindicated in pregnancy, risks involved in the use of drug clearly outweighs the benefits, (HMG CoA reductase inhibitor-statins, isotretinoin) 19 3. Describe the major pharmacokinetic (via effects on absorption, distribution, metabolism, and excretion) and pharmacodynamic (via pharmacologic [receptor] or physiologic effects) mechanisms underlying drug-drug and food-drug interactions and the potential for clinical significance of each. a. Pharmacokinetic i. Absorption: decreased motility=decreased absorption rate=lower peak plasma drug levels (doesn’t change bioavailability), increases in the rate of absorption less important clinically. Physiochemical inactivation via change in pH or formation of insoluble complexes reduced bioavailability. ii. Distribution: protein binding, displacement interactions, competitive binding may increase amount of free drug, cellular distribution interactions. Displacement of 1st drug from protein by 2nd drug results in increased levels of unbound-free 1st drug (total drug unchanged) a. Can be of clinical consequence if: i. Displaced drug has narrow therapeutic index ii. Displacing drug is started in high doses iii. Vd of the displaced drug is small iv. Response to drug occurs more rapidly than redistribution iii. Metabolism: metabolic rate increased by inducers reduced +/- subtherapeutic levels, rate decreased by inhibitorsincreased and possibly toxic levels. Most interactions occur via effects on cytochrome P450 (oxidation Phase 1 reactions) iv. Excretion: most excretion interactions occur in the kidneys and they include: 1. glomerular filtration rate a. Decreased by nephrotoxic drugs (e.g., aminoglycosides) Cp b. Increased by displacement from plasma proteins Cp 2. tubular secretion a. Decreased by competition for active transport (penicillins) Cp 3. Change in tubular reabsorption via increase in urine pH a. Decreased for weak acid drugs (e.g., aspirin) Cp b. Increased for weak base drugs (e.g., amphetamine) Cp 4. pH i. Weak Acid: R-COOH R-COO− + H+ ii. Weak Base: R-NH3+ R-NH2 + H+ 5. Change in tubular reabsorption via decrease in urine pH a. Increased for weak acid drugs Cp b. Decreased for weak base drugs Cp 6. pH i. Weak Acid: R-COOH R-COO− + H+ ii. Weak Base: R-NH3+ R-NH2 + H b. Pharmacodynamic i. Antagonistic effects: Two drugs with opposite pharmacologic effects given together 1. -blocker (hypertension) + -agonist (asthma) bronchospasm ii. Synergistic or additive therapeutic effects: Two drugs with similar therapeutic effects given together 1. -blocker + diuretic enhanced blood pressure lowering 20 iii. Synergistic of additive side effects: Similar to above, but involves side effects 1. Ethanol + benzodiazepine enhanced CNS sedation iv. Indirect pharmacodynamics effect: Pharmacologic effect of one drug indirectly affects second drug 1. Diuretic (hypokalemia) + digoxin enhanced digoxin toxicity 4. List the pharmacokinetic (decreased absorption or enhanced elimination) interventions that are available for treatment of drug overdoses and poisoning and the limitations and contraindications for each. a. Prevention of Absorption i. Emesis: empties stomach contents rapidly 1. Ipecac: emesis after 15-30 minute lag, may repeat once in 20 minutes, local irritation and CNS stimulation of chemoreceptor trigger zone (CTZ), effective orally, must be given BEFORE activated charcoal, Ipecac should no longer be used in HOME treatment 2. Apomorphine: dopamine agonist, produces emesis by stimulation of CTZ, rapid action parenterally, respiratory depressant, toxic in children, rarely used today 3. Contraindications of emetic agent: comatose pt (lacks gag reflex risk of aspiration), ingestion of corrosive poisons (strong acid/base), ingestion of CNS stimulant (risk of seizures), ingestion of petroleum distillate (risk of pneumonitis, pregnancy category C (weigh the benefit: risk, unknown if drug will cause harm). ii. Gastric lavage: most rapid and complete method to empty the stomach, but lavage + emesis only empties 30% of oral poisons. Washing of stomach contents with saline and removal via nasogastric tube, best within 60 minutes of poison ingestion. iii. Chemical adsorption Activated charcoal: binds drug in gut to limit absorption (but also binds Ipecac), effective without prior gastric emptying and can reduce elimination half-lives of drugs that have been given IV (back-diffusion of drug from blood with ion-trapping in stomach), underutilized or used in insufficient doses (best to give 10:1 ratio to toxin), serial admin may be helpful (every 4 hours), difficult to administer and poorly accepted in children, home treatment is NOT recommended. iv. Osmotic cathartics: decrease time of toxin in GI tract (osmotic laxative effect), indicated if toxin was ingested ˃60 minutes, if toxin is in enteric coated tablet or if toxin is hydrocarbon. 1. Sorbitol 70%: recommended, given with charcoal to prevent “charcoal briquet” formation 2. Magnesium citrate or sulfate: avoid in renal disease or poisonings with nephrotoxic agents 3. Sodium sulfate: avoid in CHF or HTN (system absorption fluid overload) 4. Polyethylene glycol: whole bowel irrigation that promotes elimination of entire contents of intestines, for poisonings with sustained-release drugs, metal ions, drug packets. b. Enhancement of Elimination i. Extracorporeal removal: lots of complications, pt must REALLY need this and treatment must have significantly increased rates of toxin elimination compared to normal hepatic metabolism or renal excretion 1. Hemodialysis/peritoneal dialysis: blood pumped through filter, most effective for drugs with small Vd (if large Vd, poorly removed by this method as most of drug is outside 21 plasma), toxin should have low protein binding capacity (if bound to protein, toxin won’t cross dialysis membrane), assists in correcting fluid and electrolyte imbalance 2. Hemoperfusion: blood pumped through column of adsorbent material, useful for high MW toxins with poor water solubility, risks: bleeding (removal of platelets) and electrolyte disturbances. ii. Enhanced metabolism: induction of cytochrome P450 metabolism is NOT realistic (due to 1-3 day onset of action), enhancement of detox metabolism pathways with N-acetylcysteine in acetaminophen toxicity and thiosulfate in cyanide poisoning, inhibition of metabolism to block formation of toxic metabolites (inhibition of alcohol dehydrogenase in methanol or ethylene glycol toxicity) iii. Enhanced renal excretion: previously popular but unproved value 1. Forced diuresis (fluids [normal saline] plus high efficacy diuretics [furosemide]), small effect, with danger of fluid overload, protects kidney (benefit) 2. Block reabsorption from kidney: prevention of passive reabsorption via alteration of urinary pH and ion trapping, alkalinize urine with NaHCO3 (trap weak acids pKa=3-7.5 like aspirin and barbiturates), acidify urine with NH4Cl or ascorbic acid (trap weak bases pKa=7.5-10.5 like phencyclidine or amphetamine) iv. Chelation of heavy metals: combines aspects of enhancing the elimination of the toxin (increases renal excretion) and inactivating the toxin (decreases ability to interact with and damage target tissue) 1. Heavy metal ions: ability of form coordinate covalent bonds with protein side chain nucleophiles, interact with macromolecules that are essential for normal physio function, toxin effects are due to enzyme inhibition and alteration of membrane structure. Treatment: admin of chelating agents that complex with free metal ions in body fluids reducing their concentration and promoting the dissociation of metals from these functional intracellular macromolecules, metal ion-chelator complex is excreted in the kidneys. 5. Compare and contrast the concepts of toxicokinetics (seen with toxic amounts of drugs) to “normal” pharmacokinetics (seen with therapeutic doses and therapeutic plasma levels). a. Toxicokinetics: the study of the absorption, distribution and elimination of toxic parent compounds and metabolic products that aids in prediction of amount of toxin that reaches site of injury and the resulting damage. A toxic dose of drug may result in alterations of “normal” pharmacokinetics. i. Absorption: large amount of ingested drug may slow tablet dissolution, alter GI emptying, injure GI tract altered absorption delayed peak effect ii. Volume of distribution: useful in predicting which drugs will be removed by dialysis/exchange transfution (low Vd values) iii. Clearance: important to know contribution of each organ to elimination of the toxin or drug in planning treatment strategy iv. Half-life: published values are for therapeutic doses, may be prolonged in toxic overdoses due to saturation of the elimination mechanisms 6. Describe the mechanism of acetaminophen overdose toxicity and its treatment (role of hepatic bioactivation to toxic metabolite and depleted hepatic glutathione in hepatocellular injury). 22 a. 70-80% of acetaminophen is conjugated with glucuronic acid (developed in adults) or sulfate (in children) (phase II reaction). 5-10% proceeds through a Phase I cytochrome P450 oxidation via CYP2E1 and this metabolite is the chemically reactive N–acetyl-p-benzoquinonimine (NAPQI or Ac*), a strong electrophile detoxified by phase II GSH-transferase and excreted as mercapturate. b. Hepatocellular injury occurs when there is saturation of the phase II sulfate and glucuronide conjugation pathways by toxic doses. Results in excessive formation of Ac* by the unsaturated phase I P450 pathway, eventual depletion of cellular glutathione and the binding of Ac* to critical protein or cellular constituents. Predisposing factors: increased CYP2E1 activity and decreased haptic glutathione content (occurs with excess EtOH consumption). c. Toxicity is divided into 4 stages: 1. Initial 24 hours—symptoms do NOT reflect potential seriousness (nausea, vomiting, abd pain), 2. 24-48 hours—clinical indications of hepatic damage apparent (elevated plasma aminotransferases, prothrombin time prolonged), 3. 72-96 hours—peak hepatotoxicity, 4. 7-8 days—recovery if timely treatment OR severe liver damage in 10% with 10-20% dying of liver failure. Treatment: activated charcoal and gastric lavage to remove residual drug, best within 4 hours, vigorous supportive therapy needed when intox is severe. Nacetylcysteine recommended within 12-36 hours of ingestion as this drug is through to serve as a precursor for glutathione synthesis (provides source of cysteine), administer orally (LD=140mg/kg) + 70 mg/kg every 4 hours for 17 doses, IV formulation available and decreases nausea and vomiting and has NO interference with action if emetic agent of charcoal used. 7. Describe the basic pharmacodynamic parameters of methanol and ethylene glycol that underlie their toxicities: rapid oral absorption, metabolism by common hepatic enzyme systems, these metabolic products selectively damage different tissues or organs, and toxicities of both can be treated through similar interventions. a. Methanol and ethylene glycol are well absorbed via the oral route with subsequent extensive metabolism to organic acids, have minimal toxicity until metabolized to formic acid (retinal damage and blindness) and oxalic acid (acute renal failure). The rate-limiting enzyme in the following pathway is alcohol dehydrogenase and inhibition of this enzyme (with fomepizole) is the cornerstone of treatment. Symptoms: delayed onset because need time to metabolize, serve metabolic acidosis in 4-12 hours, methanol: visual disturbances (snowstorm), ethylene glycol: deposition of calcium oxalate crystals acute renal failure. Treatment: 1. Suppress production of toxic metabolites by inhibiting alcohol dehydrogenase (infusion of EtOH to maintain a blood level of 0.1% b/c EtOH is competitive inhibitor of alcohol dehydrogenase saturates the enzyme reduces formic acid and oxalic acid) OR fomepizole, inhibitor of alcohol dehydrogenase that doesn’t produce CNS depression (more effective, more more expensive), 2. Hemodialysis, correction of metabolic acidosis 23 DD - Bacterial Structure, Function and Growth 1. Describe the major structural features of bacteria and explain the principal function(s) of each feature. b. Cell wall and cell surface structures: rigid cell wall external to plasma membrane that contains peptidoglycan. Rigidity of cell wall is essential for resisting osmotic lysis and maintaining cell shape. i. Each bacterial isolate has a characteristic, rigid shape ii. Bacterial shape is determined by both intracellular elements and by rigid components of the cell wall. Peptidoglycan layer forms a rigid mesh that surrounds the cytoplasmic membrane. It consists of a polymer with repeating units of 2 hexose sugars: N-acetylglucosamine and Nacetylmuramic acid. MurNAc residues are linked to tetrapeptide chains that contain amino acids only found in bacterial cell walls (ie: meso-diaminopimelic acid (DAP), D-glutamic acid and D-alanine). The tetrapeptides are cross-linked from one chain (via DAP in gram-negative and via L-lys in gram-positive) to D-ala on another chain. Lysozyme in body secretions contributes to innate hose defenses because it hydrolyzes the peptidoglycan by cleaving the bond between MurNAc and GlcNAc (gram-positive is MORE susceptible due to due to exposed peptidoglycan layer) Intracellular localization of each protein type iii. Other cell surface structures: 24 1. Capsules: loose, gelatinous outer surface layers and consists of complex polysaccharides. Enhance virulence by enabling the encapsulated bacteriato resist phagocytosis. Most capsular polysaccharides are antigenic, some are used as vaccines to prevent specific bacterial infections. 2. Flagella: appendages originating in cytoplasmic membrane that function as organs of motility. Peritrichous: flagella distributed all over surface. Polar: 1+ flagella at 1 end of the cell. Chemotaxis: movement toward attractive nutrients and away from toxic substances, uses flagella. Counterclockwise= swimming, clockwise= tumbling. Most flagella are antigenic. 3. Pili: aka fimbriae are long slender, proteinaceous antigenic, hair-like structures on surface of bacteria. Play a role in adherence to surfaces and tissues. Antibodies against pili may block adhernace. Sex pili: role in bacterial conjugation for ssDNA transfers. c. Cytoplasmic membrane: called the inner membrane in gram-negative, anatomical and physiological barrier between the inside and outside of the bacterial cell. Lipid bilayer made of phospholipids and proteins, but doesn’t contain sterols and has higher protein content 60-70% (compared to animal cells). i. Exhibits selective permeability: impermeable to charged substances, only hydrophobic or unchanged molecules no larger than glycerol can pass ii. Electron transport system is located on cytoplasmic membrane. Generates proton motive force during respiration. iii. Other functions: metabolite transport, biosynthesis of lipids, DNA replication and flagellar rotation. d. Cytoplasm: aqueous solution of proteins and metabolites, site of metabolism i. Ribosomes: 70S ribosomes, important for protein synthesis, Polycistronic: encode more than 1 protein product on mRNA. ii. Nucleoid: where the DNA of bacteria is located, DNA is packed and supercoiled, no nuclear membrane around the nucleoid therefore transcription and translation occur as coupled processes. Bacterial genome consists of: 1. Chromosome: single, double-stranded, circular DNA molecule, cytoskeleton as a primitive mitotic apparatus 2. Plasmids: extra-chromosomal, self-replicating DNA molecules (smaller than chromosomes), usually not essential for bacterial viability, often encode virulence factors, plasmids called “R factors” carry genes that determine resistance to antibiotics 3. Bacteriophages: viruses that infect bacteria. Temperate bacteriophages: intergrate into chromosomes and replicated as part of chromosome, usually encode for bacterial toxins, virulence factors or resistance of antibiotics. Phage conversion: change in phenotype of host bacterium as a consequence of gene expression that is encoded by a bacteriophage within the host bacterium. 2. Explain the importance of differences in cell wall structure among bacteria. a. Gram-positive: the extent of cross-linking of peptidoglycan chains is typically much greater in grampositive. Thick, extensively cross-linked peptidoglycan layer that also contains teichoic acids. Teichoic acids have a repeating polyglycerol-P or polyribitol-P backbone substituted with other molecules and 25 they are covalently attached to the peptidoglycan layer. Lipoteichoic acids are attached to underlying cytoplasmic membrane and help anchor the cell wall to membrane. b. Gram-negative: thin, sparsely cross-linked peptidoglycan layer and other major components that are located exterior to the peptidoglycan. Outer membrane is a lipid bilayer that contains lipopolysaccharide (LPS), lipoproteins and porins (transmembrane channels for diffusion)—acts as barrier to entry of some antibiotics and protects cell against detergents and toxic compounds. LPS is located exclusively in outer leaflet of outer membrane (inner leaflet consists of phospholipids). LPS contains Lipid A (toxic component of endotoxin), core polysaccharide, and O side chain oligosaccharides that function as somatic antigens (O antigen) 3. Draw a typical bacterial growth curve and explain the characteristics of each growth phase. a. Lag phase: physiologic adjustment for starting cells or inoculum. Induction of new enzymes and establishment of proper intracellular environment for optimal growth. b. Exponential (logarithmic) phase: rate of increase in cell number/cell mass is proportional to cell number/cell mass already present. A constant interval of time is required for doubling of cell number/cell mass—called the generation time (doubling time). c. Stationary phase: as essential nutrients are consumed and toxic products of metabolism accumulate, growth slows or ceases, growth that does occur is balanced with cell death. Leads to marked increase in resistance to antibiotics because antibiotics act on growing cells. In nature, bacteria are most often found at this phase. 26 d. Death phase: OPTIONAL. The number of viable bacteria decreases over time. If cell lysis (autolysis) occurs, the mass of intact bacteria in culture will decrease. 4. Describe how bacteria are classified according to their nutritional requirements a. Nutrition: provision of proper environmental conditions for promoting bacterial growth including nutrients, pH, temperature, aeration, salt concentration osmotic pressure. Under adverse nutritional conditions sporulation occurs. b. Heterotrophic: obtain carbon from an organic source c. Autotrophic: obtain carbon exclusively from CO2 d. Fastidious bacterial pathogens that are deficient in 1+ biosynthetic pathway, they require in addition to carbon and energy, a number of essential growth factors like amino acids, vitamins, purines, pyrimidines and inorganic ions. e. Obligate intracellular bacteria: grow within eukaryotic cells but cannot be cultivated on artificial media. f. Aerobe: requires oxygen to grow, produces toxic metabolites such as H2O2 and superoxide, but use catalase and SOD to protect them against ROS. g. Anaerobes: killed by oxygen, although anaerobes that are frequently associated with disease are more aeroterant than strict anaerobes. Growth Response Aerobic Anaerobic Comment Example Aerobe (strict aerobe) + - Requires oxygen; cannot ferment Mycobacterim tuberculosis Anaerob e (strict anaerobe) * - + Killed by oxygen; fermentative metabolism Clostridium sp Bacteroides sp Indifferen t (aerotolerant anaerobe) + + Ferments in presence or absence of O 2 Streptococcus pyogenes Facultative (facultative anaerobe) + + Respires with O 2; ferments in absence of O 2 Escherichia coli Staphylococcus aureus (+) + Grows best at low O 2 concentrations; can grow withou t O2 Campylobacter jejuni Type o f Bacteria Microaerophilic ( + ) indicates small amoun t of growth 27 5. Define respiration and fermentation and explain how metabolic “energy currency” is generated. a. “Energy currency” in bacteria: ATP and electrochemical gradients (proton motive force), these 2 types of potential energy are interconvertible by the membrane ATPase. b. Fermentation: organic compounds serve as both electro donors are electron acceptors (no net oxidation of substrates occurs). Anaerobic and facultative bacteria grown under anaerobic conditions obtain energy by fermenting organic substrates. Indifferent organisms obtain energy by fermentation under anaerobic and aerobic conditions because they are incapable of respiration. c. Respiration: generate ATP through electron transport and use molecule oxygen as final electron acceptor, anaerobic respiration—certain bacteria use inorganic substrates such as nitrate as terminal electron acceptors instead of O2. 6. Explain why unique bacterial components are important as potential targets for antimicrobial therapy. a. Selective toxicity: selective inhibition of microbial growth at drug concentrations tolerated by the hose. Some components of bacteria that are NOT present in eukaryotes or are sufficiently different from their counterparts to be effective targets for antimicrobial agents. 7. Identify the principal targets for the major groups of antibiotics used in human medicine. a. Cell wall-active microbials: selective toxicity due to lack of peptidoglycan in mammalian cells i. Beta-lactams, vancomycin, cycloserine b. Outer and cytoplasmic membrane-active microbials: polymyxins are cationic surfactants that disrupt bacterial outer and cytoplasmic membranes, they are less active on mammalian cell membranes c. Inhibitors of protein synthesis at the ribosomal level due to the differences between bacterial and mammalian ribosomes i. Aminoglycosides, tetracyclines, chloramphenicol, macrolides/lincomycins d. Inhibitors of nucleic acid synthesis i. Quinolones, rifampicin e. Metabolic inhibitory antimicrobials i. Sulfonamides, trimethoprime, isoniazid, metronidazole 28 DD LAB- Principles of Bacterial Isolation and Identification 1. To isolate the individual pathogenic organism from a mixed culture and identify the individual bacteria by applying basic microbiological techniques such as Gram stain, biochemical properties and serological testing 2. To identify a pathogenic organism from a simulated urine sample, to illustrate how many bacteria must be present in samples to detect them by microscopy. 3. Understand differential and selective media including identifying which media is differential or selective and how these media are used in microbiology. DD - Host-microbe interactions a. Define and describe a. Infection: process whereby a microbe enters into a relationship with the host. It may or may not cause disease. b. infectious disease: disease caused by an infection with a microbe. Some infections are communicable (transmitted patient to patient) and others are not. c. Pathogenicity: ability (usually of a species) to cause disease i. Frank pathogens: causes disease readily in normal hosts ii. Opportunistic pathogens: cause disease in compromised hosts, many normal flora are opportunistic pathogens d. Virulence: denotes degree of pathogenicity. If a strain is highly virulent, then it is likely to cause disase when introduced to a host in small numbers 2. Explain how a microbe is shown to be the cause of a specific disease a. Use Koch’s Postulates: i. Specific microbes are present regularly in characteristic lesions of the disease ii. Specific microbes can be isolated and grown in vitro iii. Injection of cultured microbes into animals reproduces the disease seen in humans iv. Specific microbes can be re-isolated from lesions of the disease in animals v. LIMITATIONS: 1. Some infectious diseases do not have a characteristic (pathognomonic) lesion 2. Some microbes cause specific infectious diseases but cannot be grown in vitro 3. Traditional concepts of pathogenicity focus primarily on properties of microbes vs. hosts 4. The characteristics of infectious diseases usually reflect complex interactions between microbes and their hosts. 3. Describe typical stages in pathogenesis of an infectious disease and explain their importance a. Encounter: how the agent meets the host i. Endogenously (normal flora) or exogenously (from environment), route of infection, what is infectious dose b. Entry: how the agent enters the host i. Did microbe cross epithelial barrier (by invasion or passively?) ii. Colonization of body surfaces iii. Adherence c. Spread: how the agent spreads from the site of entry 29 i. Spreading factors: hyaluronidase, elastase, collagenase ii. “wall off” infection: coagulase d. Multiplication: how the agent multiples in the host i. Must replicate at levels that exceed their clearance by the host. e. Damage: how tissue damage is caused by the agent and/or the host response i. Aggressins: microbial products that damage the host ii. Impedins: microbial products that block host defenses f. Outcome: dose the agent or host win the battle or do they learn to coexist g. Mechanisms of host response to infection 4. Compare mechanisms of innate and acquired host defense against infections 5. Describe the composition and importance of the normal flora of the body a. Factors that influence normal flora i. Diet (breast-feeding, bottle feeding, solid food) ii. Suppression of flora with antibiotics iii. Anatomic abnormalities (blind loop increase flora) iv. Genetic differences between individuals b. Physiologic importance of normal flora i. Effects on tissue/organ differentiation (normal vs. germfree animals) ii. Production of vitamins by gut flora iii. Biochemical conversions iv. Competition with pathogens for colonization of body surface 30 6. Compare several disease paradigms that illustrate selected mechanisms of pathogenesis a. Cholera: toxin mediated disease, organism is non-invasive b. Pneumococcal pneumonia: acute inflammation caused by invasive extracellular bacteria, invades and replicates c. Tuberculosis: infection by a facultative intracellular bacterium, grows in phagocytes d. Rheumatic fever: pathology mediated by an immune response, from streptococcal infections DD - Microbial Toxins 1. Define and describe the term “microbial toxin”. a. Macromolecular products of microbes that cause harm to susceptible animals by altering cellular structure or function, they are very potent, the clostridian neurotoxins are the most toxic biological substances known. Importance: some toxins cause the major manifestations of specific diseases, other toxins contribute to pathogenesis without causing unique signs or symptoms, toxin-mediated diseases cause significant morbidity and mortality 2. Explain how a microbial toxin is implicated in pathogenesis of an infectious disease e. Show that purified toxin causes the same symptoms or signs as infection by the toxin-producing microbe f. Show that antitoxin prevents disease caused by the toxin producing microbe g. Show that virulence of individual bacterial strains correlates with the amount of toxin that they produce h. Show the nontoxinogenic mutants are avirulant and that virulence is restored if the microbe regains the ability to produce toxin. 31 3. Explain the mechanisms of action of the microbial toxins described here and compare the properties of microbial toxins that have different mechanisms of action a. toxins that facilitate spread of microbes through tissues: toxic enzymes break down ECM or degrade debris in necrotic tissue b. toxins that damage cellular membranes: called hemolysins (easy to see action of RBC), but act on other cells too, therefore cytolysins is accurate name. These toxins insert into membrane, assemble into complexes that form forms and lyse the target cell. c. toxins that stimulate cytokine production: pyrogenic exotoxins are from a larger class of molecules called superantigens—the most potent known T cell activators (bind to both MHC class II molecules and to V-beta chains on T cells and they activate a larger number of T cells than any specific antigen does. d. toxins that inhibit protein synthesis: diphtheria toxin and Pseudomonas aeruginosa exotoxin A inactivate elongation factor 2 (required for peptide chain elongation) Shiga toxins and ricin (plant toxin) remove one adenine residue from RNA of ribosome to inactivate it. e. toxins that modify intracellular signaling pathways: Heat-labile enterotoxins of Vibrio cholera and E. coli increase cell membranes associated cAMP, activate alpha subunit of stimulatory Gs regulatory protein active chloride secretion and secretory diarrhea. Pertussis toxins: increase cAMP and inactivate the alpha subunit on inhibitory Gi regulatory protein. f. toxins that inhibit release of neurotransmitters 4. Explain the principles of immunization against toxin-mediated diseases. a. Antitoxin antibodies (antitoxins) bind to toxins and prevent their toxicity (neutralization). Antitoxins don’t prevent infection by toxin-producting bacteria or reverse toxic effects after toxin has entered host cells. b. Toxoids are derivatives of toxins that retain immunogenicity but lack toxicity, they are used as vaccines for long term protection c. passive immunization is the administration of antibodies to a patient to provide immediate but temporary protection against a toxin or infectious disease d. active immunization involves the administration of toxoid to a patient in order to elicit production of specific anti-toxic antibodies. Primary series of immunizations and periodic boosters required, active immunity can persist for years because of immunologic memory. 5. Explain the principles for developing novel therapeutic agents based on toxins. Immunotoxins are hybrid molecules which contain the active (A) fragment of a toxin (e.g., diphtheria toxin, exotoxin A, ricin, etc.) chemically conjugated to, or expressed as a fusion protein with, ligands (e.g. monoclonal antibodies, "single-chain" antibodies, or the receptor-binding domains of hormones) for specific receptors that differ from the receptors for the native toxins. The rationale is to eliminate the receptor-binding component of the native toxin and provide a new receptor-binding moiety that will redirect the toxic component to target cells that express the alternative receptor. Many immunotoxins are designed to kill tumor cells that display a tumor-specific receptor but not normal cells that lack that receptor. Immunotoxins are being tested as potentially valuable therapeutic agents for treatment of specific cancers, autoimmune diseases, and other disorders. 32 DD - Genetic Variation, Gene Transfer and Evolution of Virulence a. Describe the mechanisms that generate genetic diversity within a bacterial species and how these contribute to the evolution of virulence a. Spontaneous mutation: single base changes, deletions and insertions. Mutation rates are very low: 10-610-10 per cell-generation. Examples: increased resistance to antimicrobials in Pseudomonas and Mycobacterium tuberculosis. b. Recombination: either site-specific or homologous recombination within a particular organism or genetic exchange and recombination between closely related organisms i. Example: recombination between variant pilin genes produces hybrid genes that encode pilin with new unique antigenic properties c. Acquisition of new DNA segments: lateral transfer from other bacteria, even from unrelated species. New genes may alter virulence potential, survival characteristics, antimicrobial resistance i. Acquisition of transposable elements—a discrete segement of DNA which is capable of moving itself from one chromosomal location to a new location within the cell. Typically encode 1+ proteins that mediates transposition (transposase). 1. Insertion sequences: transposons that only encode transposase 2. Complex transposons: carry additional genes such as those encoding antibiotic resistance, toxins, adhesions, etc. ii. Bacteriophage conversion: certain virulence genes are carried on bacteriophage and are not a “normal” component of the respective bacterial genome. iii. Acquisition of plasmids: can be transferred from one bacteria to another by conjugation or transduction. They can carry virulence genes or genes conferring resistance to antibiotics iv. Acquisition of pathogenicity islands: Pathogenicity islands are large segments of DNA present in the chromosome of some, but not all strains of bacteria. The encode genes that contribute to the virulence of these isolates. Lacking PI may make bacteria avirulent. b. Discuss how spontaneous mutation and selection can interact to determine the genetic composition of bacterial populations a. Errors occur that result in base pair changes, deletions, duplications, etc. b. Typically changes are deleterious or neutral c. In rare instances, a mutation may confer a selective advantage d. Spontaneous mutation to antibiotic resistance occurs once in approx 108 – 1010 organisms c. Distinguish between transformation, transduction and conjugation as mechanisms of gene transfer. Identify the salient features of each mechanism a. Transformation: first to be discovered, active component in transformation is naked DNA (chromosome fragment or plasmids), many transformable species only become competent for DNA uptake at certain points in their growth cycle, occurs most frequently between members in same species. b. Transduction: gene transfer mediated by a bacteriophage, bacterial virus (bacteriophage) transfers segments of DNA from 1 cell to another. c. Conjugation: genetic transfer that is dependent upon physical contact between the donor and recipient, mediated by bacterial plasmids 33 d. e. d. Discuss the properties of bacterial viruses. Distinguish between the lytic and lysogenic state. a. Lytic: phage multiples and host cell is lysed. b. Lysogenic: host cell remains viable and infecting phage DNA is maintained by the host cell in a noninfectious state called prophage. e. 34 f. Describe how errors in bacteriophage development can lead to phage-mediated gene transfer a. During phage assembly, if there is any error in DNA packaging and the packaging system inserts a “headful” sized piece of bacterial DNA into a maturing phage capsid in place of a normal phage DNA molecule then the transducing particles contain no viral genetic information, but they are still able to attach to other host cells and inject the bacterial DNA which they contain. Injected DNA may then recombine with homologous segement in recipient to produce a genetic recombinant, transductant. g. Define lysogenic conversion. Distinguish between lysogenic conversion and generalized transduction a. Lysogenic conversion: certain temperate bacteriophage encode genes which may be expressed during the lysogenic state and cause the appearance of a new phenotypic trait in the lysogenic host. This is called bacteriophage conversion or lysogenic conversion. The genes controlling the new phenotypic trait are found only as a component of the phage genome (the converting genes are NOT found alone as normal constituents of the bacterial genome). Examples: diphtheria toxin, scarlet fever toxin, cholera toxin. b. Generalized transduction: process in which any segment of the donor cell genome (chromosome or resident plasmids) may be passed into another cell. 35 DD - Common Bacterial Pathogens Genus Bacteria Staphyl ococcus Staphylo coccus aureus Appearanc e (micro) blue/purpl e cocci in clusters, GRAPES Staphyl ococcus Staphylo coccus epidermi dis Blue cocci Streptoc occus Streptoc occus pyogenes Blue cocci in chains of pairs Gram Positive (+) Cocci Disease General Primary pathogenic species in genus. Asymptomatic in 30% of healthy ppl. Site of carriage: anterior nares, perineum. Endogenous flora can lead to infection of self or others (ie: healthcare worker transmits to patient). Catalase +. Prototype of SSNA’s (staph species, not aureus) or CNS (coagulase – staph). Considered normal skin flora/non-pathogenic, but can cause localized infection usually associated with foreign body (catherter, shunts, prostheses). Catalase +. Catalase (-). “Group A Strep” of which there are 80 serotypes (antigenic differences of M protein). 36 Treatment/Drugs Typical disease: 1. Cutaneous infection (boils, folliculitis, wound infection), characteristic lesion is localized abscess. Bacteria + host help form fibrinous capsule that walls off infection and limits spread, but limits access of phagocytic cells, antibodies, etc. “coagulase” is virulence factor that forms fibrin capsule and deposits fibrin on cell surface. Cutaneous staph associated with foreign body (suture/splinter) that interferes with bacterial clearance by phagocytes + provides surface for bacteria to grow. 2. toxin-mediated disease, certain strains have genes for 1+ protein toxins. Scalded skin syndrome (exfoliatins) Local infection, toxin production (epidermolytic toxins A and B), systemic effects. Widespread desquamation in infants; localized in older. Toxic shock syndrome (toxic shock syndrome toxins produced and circulate leading to systemic effects) and staph food poisoning (staph enterotoxins via ingestion of contaminated food). 3. pneumonia: pts with impaired host defenses at risk “slime” production, an extracellular glycocalyz that allows organisms to adhere very tenaciously to implanted device and allows them to grow in a protected biofilm on surface. Drain abscess. 1. Pharyngeal Infection: Cause of “strep throat”. Virulence factor “M” protein is a surface exposed protein that inhibits phagocytosis Untreated is self-limiting weeks. Production of Mantibody by host makes b killing and leads to recove DRUGS: Pen G/Pen V if PCNase-producing (MSS DICLOX, 1st CEPH if CA-MRSA: TMP-SMX, TC If HA-MRSA: VANC, LIN, D Difficult to treat, require Antibiotic resistance (incl limited access of drug to Transmission: nasal secretions or by droplets from coughing. and killing by PMN’s and increases adherence to epithelial cells. 2. Skin and wound infection: typical lesion is that of a spreading infection of cutaneous/subQ tissue. Bacteremia/sepsis possible. Infection uses hydrolytic enzymes to break down tissue and damage/kill phagocytic cells (facilitates spread of organism). Streptoc occus Streptoc occus pneumon iae Streptoc occu Enteroco ccus faecalis Blue cocci in pairs, diplococcic, “pneumoc occus” Normal flora in UR tract of 40% of healthy people. Predisposing factors: young/old, alcoholism, respiratory viral infection Primary cause of enterococcal infections. Infection sites: urinary tract, surgical wounds, biliary 37 3. Post-streptococcal diseases: a) Glomerulonephritis: immune complex disease that may follow skin or pharyngeal infection by Group A strep. Streptococcal antigen-antibody complexes deposited in kidney and accumulate. Self-limiting. Complete-mediated damage to kidney. b) Rheumatic fever: autoimmune inflammatory disease that may follow group A strep pharyngitis (this is why we treat). Symptoms: fever, inflamm heart, joints. Antibodies produced during response to pharyngeal infection self-react (only from rheumatogenic strains) are recognize and bind specific host antigens in myocardium and heart valves. NOT like endocarditis (a true bacterial infection of heart values) because there are no bacterial colonies . 1. Pneumonia 2. Meningitis 3. Bacteremia 4. Sinusitis, otitis media, bronchitis Pathogenesis: related to ability ot grow and evade host defenses, antiphagocytic polysaccharide CAPSULE (91 distinct antigenic capsules), recovery/immunity due to anticapsular antibody 1. Frequent cause of nosocomial (acquired IN-hospital) infections, transferred on hands of hospital staff. Bacteremia, meningitis, UTI. antigenically related strai DRUGS: PEN G/PEN V AMOX MAC 1st CEPH VANC Vaccination: 23-valent va 13- valent in children. Eff "serotype replacement” DRUGS: PEN G/PEN V MAC FQ If resistant, 3rd CEPH DRUGS: VANC CARB PEN G/AMP +/- AG Gram Positive (+) Rods Genus Bacteria Appearanc e Clostridi um Clostridiu m difficile Blue rods tract. Normal flora in GI tract of some people. Often seen as mixed infection of several different organisms, including anaerobes (important in cases of perforated colon and release of contents) General Diseases Treat Diseases associated with antibiotic treatment for unrelated conditions (first described with use of clindamycin). Caused by depletion of intestinal flora by antibioticovergrowth of C. difficile (self bacteria or transmitted from another patient). Bacteria produces 2 discrete toxins that damage mucosa of intestines. 1. Diarrhea 2. Pseudomembranous colitis 1. Tetanus, toxin-mediated. Local infection (anaerobic) and toxin production Retrograde transport of toxin to CNS Blocks inhibitory interneurons in CNS Vaccine vs antitoxin Resis antib DRUG METR VANC Fidax Strict anaerobes—killed by O2 Endospore formers (Critical to persistence of these bacteria) Clostridia most likely to be encountered in hospitalnosocomial infections. Normal flora of GI tract in 10% of healthy ppl. Clostridiu m tetani Soil and animals (importance of spores) Clostridiu m botulinu m Soil and animals (importance of spores) Clostribiu m perfringe ns Gram Negative (-) Rods Escherich “typical” ia coli gram (-) rod If VRE—LIN/SG DRUG CLIN 1. Botulism, toxin-mediated Preformed toxin in food Toxin blocks acetylcholine transmission at neuromuscular junctions Part of normal flora in human GI tract (large intestine) 38 1. Gangrene/tissue infections DRUG CLIN Disease caused by endogenous organisms or acquisition (ingestion) 1. GI disease: drinking contaminated H2O, ETEC (enterotoxigenic E. coli, traveler’s diarrhea). Bacterial properties: adherence to intestinal mucosa (pili) and toxins that disrupt electrolyte balance in gut. Selflimiting. 2. UTI: typically endogenous from GI tract, access the UT via urethra—bladder—kidney. “special” strains get into “wrong” place. Bacteria properties: adherence to bladder epithelium, specific interactions with bladder epithelial cells, hemolytic. 3. ABD infection: escape of contents of colon to 1. Flu DRUG FQ TMPAMO 1st CE AG NF Pseudom onas aerugino sa Gram Negative (-) (Diplo)cocci Neisseria gonorrho eae Very common environmental bacterium, most ppl highly resistant to infection Causative agent of gonorrhea and conjunctivitis blindness in infants born to infected mothers peritoneal cavity, surgical/trauma wounds, colon cancer. Bacteriologically mixed cultures. Associated with anaerobic bacteria to form anaerobic abscess. Opportunistic pathogen, infection of burns, wounds. Seen in immune compromised patients. 1. infections of traumatic injury, surgical wounds and especially BURNS 2. Chronic lung infection of patients with CF: S. aureus infections are common early in life (viscous secretions statsis in lungs infection), but treated with antimicrobials. By age 15-20 CF pts become chronically infected with P. aeruginosa (intrinstic resistance to many anti-staph drugs and protected from phagocytosis by viscous lung secretions/mucois exopolysaccharide made by bacteria) and progressive lung damage due to action of toxins and host immune response begins frequent cause of death in CF pts Key to infectivity is pilus: required for virulence, adherence and interferes with bacterial killing by neutrophils. Different strains have antigenically distinct pili and bacterial can undergo process of antigenic variation during infection (cells switch to distinct type of pili). Means pt can have repeated infection. Growth on mucosal surfaceinflamm responsepurulent discharge/local tissue invasion. Prolonged infection=scarring and fibrosis. Males: asymptomatic to urethritis Females: ˃rate of asymptomatic is higher in females, infection of cervix, urethra, ascends to uterine tubesfibrosis and infertility. Diffic patie resist to ma diffic drugs replic DRUG FQ, C Antib prote Asym easily becau they DRUG CEFT FQ MAC Anaerobic Bacteria Most common anaerobic bacteria (besides Clostridia) are members of normal flora that inhabit colon, mouth (gums/teeth), female Killed in presence of molecular O2. Diverse gram staining/morphology. Common properties: generally of endogenous origin (norm typical lesion: abscess (just like Staph aureus). Hallmark: mixed infection containing aerobic and anaerobic bacteria (aerobes metab conducive to anaerobe growth). Because of anaerobic metabolism some drugs are more effective (metronidazole) and some less (a Obligate intracellular bacteria: grow only in infected eukaryotic cell (Cannot be Cultured) Rickettsia 1. Rocky Mountain spotted fever, lost capacity to Drug synthesize own ATP (rely on host cell) cell e DRUG DOXY CHLO Chlamydi Obligate intracellular 1. Trachoma: chronic conjunctiva infection Drug a bacteria scarring/blindness, endemic in Asia and Africa where cell e poor hygiene prevalent 2. Genital: common STD, found with coinfections of N. DRUG gonorrhoeae. Causes “non-gonococcal urethritis” in TCN men and urethritis, cervicitis and PID in women. MAC 3. Neonatal: if mom has disease, baby can be infected at birthconjunctivitis and pneumonia. 39 Bacteria without cell walls and containing sterols in plasma membrane Mycopla Mycoplas Person-to-person (infected respiratory sma ma secretions). Organism grows in specialized pneumon cell-free bacteriologic medium, but is iae difficult/slow. DX:serological test, gram stain rules out other bacterial causes. 40 1. Common cause of pneumonia in ages 5-20. Generally mild w/fever, HA, sore throat, nonproductive cough, aches, fatigue. Recovery in 14 weeks. Adheres to respiratory epithelial cells, growth is extracellular, bacteria produce H2O2 and superoxide radicals damages host tissue. Becau shape pleom are n DRUG MAC Antibacterial Agents I-III Define and / or give examples for: 1. Selective Toxicity a. Effects of antimicrobial agents should be exerted selectively on the MICROBE, NOT the host. Fundamental feature of antibiotic therapy. No perfect antibiotics exist. b. Biochemical differences between pathogen TARGET and host must be discovered and exploited. i. Inhibition of metabolic pathway found in bacteria, NOT humans 1. Folate metabolism: bacteria make folate intracellularly, humans take up folate from environment ii. Pathway exists in bacteria and humans, but differences in enzyme structure 1. Protein synthesis: bacterial ribosome is 30S and 50S, mammals use 40S and 60S 2. Nucleic acid synthesis: DNA gyrate (bacteria) vs. topoisomerase (humans). RNA polymerase is structurally distinct in bacteria too. iii. Macromolecular structure does NOT exist in humans 1. Cell wall synthesis: peptidoglycan component does not occur in eukaryotes. iv. Macromolecular structure differs between microbes and humans 1. Fungal cell membrane: ergosterol is major constituent f fungal membranes vs. cholesterol in mammals. 2. Antibacterial Spectrum: Narrow vs extended vs broad a. Narrow spectrum: effective against either gram positive OR gram negative), most effective on susceptible organisms, less disturbance on host flora. i. Animoglycosides, bacitracin, clindamycin, vancomycin, metronidazole, Pen G, Pen V, Penicillinase-resistant penicillins, monobactams b. Extended spectrum: effective against gram positive AND gram negative i. Extended-spectrum penicillins, cephalosporins, fluoroquinolones, carbapenems c. Broad spectrum: effective against gram positive, gram negative and atypical, sacrifice efficacy for greater scope of activity for initial empiric coverage, less likely to cause superinfections, acute severe infections should be treated with broad spectrum (target empiric therapy to likely bacteria) with a switch to narrow spectrum ASAP (target definitive therapy to bacteria based on lab results). i. Macrolides, chloramphenicol, sulfonamides, tetracyclines, trimethoprim 3. Resistance: Natural vs escape vs acquired (chromosomal vs plasmid-mediated) a. Natural (intrinsic) therapy: when a bacteria naturally doesn’t have a susceptible target for drug action. Example: fungal cell walls don’t have peptidoglycans and mycoplasma don’t have cell walls at all, therefore they are naturally resistant to penicillins which interfere will cell wall production b. Escape: microbe is sensitive and antibiotic reaches target but organism escapes the consequences dueto availability of purines, thymidine, serine, methionine released from purulent infections (sulfonamide resistance) or failure to lyse due to lack of osmotic pressure difference (penicillin resistance), emphasizes important role of surgical drainage if needed. c. Acquired resistance: selective pressure (ie: antibiotic administration) successive generations of organisms with biochemical traits that minimize drug action i. Mutational (chromosomal) resistance: rate is 1 in 10^7-10^12. Each generation becomes more resistant if allow to survive, thus proper dosing and duration of antibiotic therapy prevents survival of slightly resistant strains. 41 ii. Plasmid mediated resistance: clinically important source of multiple drug resistance that can emerge during a SINGLE course of treatment. Nonpathogenic coliform bacteria can code for resistance to multiple drugs via a protein that moves antibiotics out of cell. Exchange of genetic information occurs via: 1. Conjugation: 2 physically attached bacteria, exchange of plasmid w/ resistant determinant 2. Transformation: ability of bacteria to pick up free DNA from environment 3. Transduction: with virus (bacteriophage) carrying resistance determinant R to bacteria. 4. Mechanisms of resistance and implications for therapy a. Altered targets or receptors i. Penicillin-binding proteins [MRSA, S. pneumoniae, Enterococci) 1. -lactam antibiotics (penicillins, cephalosporins, carbapenems) ii. DNA gyrase [S. aureus, Pseudomonas species] 1. Fluoroquinolones iii. Peptidoglycan side chain [Enterococci (VRE), Staphylococci (VRSA)] 1. Vancomycin iv. 50S ribosome methylation [Streptococci, Staphylococci, Enterococci] 1. Erythromycin, Clindamycin b. Enzymatic destruction or inactivation of antibiotic i. -lactamase [S. aureus, P. aeruginosa, Bacteroides, Enterococci] 1. -lactams (penicillins, cephalosporins) ii. Acetyl-/phospho-/adenylyltransferases [Enterococci] 1. Aminoglycosides iii. Acetyltransferase [Staphylococci, Streptococci, Neisseria] 1. Chloramphenicol c. Alternative resistant metabolic pathway i. Overproduction of PABA or thymidine nucleotides [Streptococci] 1. Sulfonamides d. Decreased entry (natural resistance) i. Pseudomonas aeruginosa 1. -lactam antibiotics ii. Pseudomonas species 1. Fluoroquinolones iii. E. coli, Pseudomonas 1. Aminoglycosides e. Increased efflux (MDR may be encoded by single gene) i. Streptococci, Staphylococci, Enterococci 1. Tetracyclines, Macrolides ii. Pseudomonas species 1. Fluoroquinolones f. Resistance can be minimized by only using antibiotics when need is established, selecting antibiotic on basis of susceptibility tests, using adequate concentration and duration to prevent first and second step mutants. 42 5. Describe: a. Classifications of antimicrobial mechanisms of action i. Inhibitions of synthesis or damage to cell wall ii. Inhibition of synthesis or damage to cell membrane iii. Modification of synthesis or metabolism of nucleic acids iv. Inhibition or modification of protein synthesis v. Modification of intermediary metabolism (folate metabolism) b. Which mechanisms generally result in bacteriostatic or bactericidal effects i. Whether antibiotic has –cidal or –static action is determined by its mechanim of action (TARGET), concentration achieved in vivo and the specific microorganism. ii. Bacteriostatic: organisms that are prevented from growing 1. Mechanism: inhibition of protein synthesis (exception are aminoglycosides, which are –cidal), inhibition of intermediary metabolic pathways. iii. Bactericidal: organisms are killed 43 1. Mechanism: inhibition of cell wall synthesis, disruption of cell membrane function, interference with DNA function or synthesis c. Advantages of bactericidal agents i. Preferred in severe infection ii. Act more quickly and their action is often irreversible iii. Compensate for patients with an impaired host defense iv. Required for treatment of infections in locations that are not accessible to host immune system responses d. Importance of pharmacokinetic and host factors in selection of antimicrobial therapy i. Absorption: necessary to achieve adequate concentration in systemic circulation, accomplished via oral or IV route, some infections can be treated with topicals (to skin or mucous membranes) or non-absorbable drugs for tx of GI tract infections. 1. Oral route advantages: easy to administer, patient acceptance, lower cost. 2. Oral route disadvantages: GI upset, diarrhea, alter natural flora, incomplete absorption, unsuitable for NPO pts. a. Take on empty stomach: when antibiotic is unstable to increased acidity that occurs when food is in the stomach b. Take with food or meal: when drug is stable but may be irritating to stomach 3. IV route advantages: most rapid, predictable plasma levels 4. IV route disadvantages: greater training needed, greater expense, aseptic technique required 5. Typically, patients are switched to oral antibiotics ASAP from IV (to reduce $ and complications) ii. Distribution: 1. CNS: most antibiotics distribute well to tissues outside of CNS, but vary in ability to cross BBB. a. Readily enter CSF: chloramphenicol, sulfonamides-trimethoprim, cephalosporins (3rd/4th), rifampin-metronidazole b. Enter with inflammation: penicillins, vancomycin, ciprofloxacin, tetracycline 44 c. Enter CSF poorly: aminoglycosides, cephalosporins (1st/2nd), erythromycin, clindamycin 2. Selective distribution (accumulation): a. Beneficial accumulation: clindamycin into bone (tx osteomyelitis), macrolides into pulmonary cells (tx URI), tetracyclines into gingical crevicular fluid and sebum (tx peridntitis and acne), excretion of nitrofurantoin into urine (tx UTI) b. Increased potential for toxicity accumulation: aminoglycoside binding to cells of inner ear and renal brush border ototoxicity and nephrotoxicity, tetracyclines binding to Ca++ in developing bone and teeth abnormal bone growth and brown tooth discoloration in children. 3. Fetus: antibiotics can have adverse effects on fetus if they cross placenta. Drugs to use with caution during pregnancy: aminoglycosides, metronidazole, chloramphenicol, tetracyclines, fluoroquinolones, voriconazole iii. Elimination 1. Renal excretion: renal dosing (dose +/- frequency of antibiotic is adjusted based on renal function) may be necessary for pt with kidney dysfunction. Renal status monitored with serum creatinine (Scr) and creatinine clearance (CrCl) 2. Hepatic metabolism: metabolic drug-drug interactions, interpatient differences in metabolic rate (genetic polymorphisms), hepatotoxic antibiotic actions. No lab value to give estimate of liver’s ability to metabolize antibiotics. Generally, heptatically metabolized antibiotics are avoided in pt with liver dysfunction 3. Duration of antimicrobial activity: half life information helps to guide dosage regimen a. Too short or dose too low: resistance can develop, reccurance of infection b. Too long: superinfection more likely c. Too high: dose-related toxicities d. Post-antibiotic effect: some anitbiotics continue to kill/inhibit bacterial growth for several hours after [drug] falls below MIC, can be given less frequently e. Concentration-dependent killing: antibiotics that kill bacteria faster when given in doses that result in higher plasma concentrations 6. For the following drugs and drug categories (listed below) describe their: General mechanism of action (include discussion of -cidal vs -static, mechanisms of resistance) Pharmacokinetics: Absorption (oral vs parenteral) / Distribution (esp., CNS penetration) / Elimination (renal vs hepatic) Spectrum with respect to the following broad bacterial classifications (bolded) and Clinical Uses (underlined) as related to common causative organisms (italics) COCCI a. Penicillin: i. Substitutions on R group to increase acid stability (in GI), decrease renal excretion, increase metabolic stability, minimize bacterial resistance, increase antibacterial spectrum by increasing bacterial penetration 45 ii. Mode of Action 1. Inhibit Bacterial Cell Wall Synthesis at Stage 3: a. Stage 1: Synthesis-assembly of cell wall subunits occurring in the cytosol (inhibited by fosfomycin and cycloserine. b. Stage 2: Linear polymerization of subunits occurring at cell membrane (inhibited by bacitracin & vancomycin). Target transglycosylase. c. Stage 3: Cross-linking of peptidoglycan polymers occurring at the cell wall (inhibited by penicillins, cephalosporins. Target transpeptidase, carboxypeptidase (PBPs) 2. Penicillins are bactericidal to growing organisms, Pen G confined to gram + organisms and gram – cocci. 3. Penicillin Binding Proteins: Beta-lactam antibiotics acylate several bacterial proteins termed penicillin binding proteins (PBP’s). PBPs include (but aren’t limited to) transpeptidase enzymes. Penicillin’s inhibit these enzymes by irreversible covalent interaction. Certain beta-lactam antibiotics bind to distinct PBPs. Binding is not uniform. Beta-lactams trigger autolytic activity via presence of endogenous autolytic enzymes by depressing the natural inhibitory action of autolysins. Effect persists when drug is gone due to penicillin’s covalent binding to bacterial proteins. Maximal killing is function of growth rate of organism. 4. Resistance: a. Production of penicillinase enzyme via plasmid. Production of beta-lactamase induced in presence of penicillin. Transmitted to sensitive organisms by bacteriophages. Major problem with staphylococcus. b. Alterations in penicillin binding proteins, responsible for methicillin resistance in staph (MRSA) and penicillin resistance in pneumococci. c. Inability to penetrate bacterial cell d. Metabolically inactive organisms or “L” forms can survive in hypertonic environment like kidney. iii. Pharmacokinetics 1. Absorption: moderately strong acids, highly water-soluble, acid-lability impairs oral absorption of many penicillins and optimal absorption is on empty stomach. Chemical modification of R group improves absorption by increasing acid stability. Rapidly absorbed from IM parenteral sites. 46 2. Distribution: throughout body, penetrates tissues poorly (ionized at physiological pH), high concentration in liver, kidney, skin. Can enter inflamed tissues or membranes (CSF, joint, eye) more readily than normal. 3. Metabolism-Excretion: most excreted as active drug via kidney, 1 hour half-life, 90% by tubular secretion, blocked by probenecid, metabolism increases in cases of renal failure, excreted in break milk (consider risk:benefit) iv. Toxicity/Adverse reactions 1. Virtually non-toxic, except for hypersensitivity reactions a. Type I reaction: very rare (0.05%) but life threatening, mediated by IgE (mast cells) antibodies, onset: few minutes- 30 minutes. Symptoms: urticarial, angioedema, respiratory obstruction, vascular collapse. b. Type II reaction: rare, due to cytotoxic antibodies of IgG or IgM class, complement-dependent cell destruction (hemolytic anemia). c. Type III reaction delayed (˃72 hours), formation of IgG or IgM immune complexes with penicillin that act as antigens and can activate complement and lodge in tissue skin rash, serum sickness, arthralgias, allergic vasculitis d. Type IV reaction: cell mediated allergy, delayed reactions, mediated by Tlymphocytes, skin eruptions and thrombocytopenia. e. Other: idiopathic, most common reaction (1-4%) is maculopapular or morbilliform rash (mild and reversible) v. Individual Penicillins 1. Prototype Penicillin (narrow spectrum of antimicrobial activity) a. Penicillin G: prototypical, powerful, inexpensive, penicillin of choice in most cases, disadvantages: hydrolyzed by acid and penicillinase enzyme. 30-50% bound to plasma protein. Most common use is via parenteral route to pts with serious infections. b. Penicillin V: acid resistant, better absorbed than Pen G, but still incompletely. Pen V is preferred for oral therapy due to higher reliability of absorption. Antimicrobial efficacy ˂ that of Pen G, but suitable for mild-mod infections. 2. Penicillinase-resistant penicillin a. In order of efficacy (greatest first): Methicillin (obsolete) ˃ Nafcillin (parenteral, erratic oral) ˃ Oxacillin (oral) ˃ Cloxacillin (oral) and Dicloxacillin (oral). b. These antibiotics are ˂ potent against Pen G-sensitive organisms and are not subsitutes for Pen G except when penicillinase-producing organisms are present. c. Acid resistance varies among group members, these penicillins are less susceptibe to beta-lactamase than cephalosporins. d. Eliminated by both renal and hepatic routes e. All are narrow spectrum: gran +, gram – cocci. 3. Extended spectrum penicillin a. Increased hydrophilicity allowing penetration through porins of outer membrane of gram – organisms b. Ampicillin and amoxicillin have additional activity against gram – bacilli. Acid resistant, but not resistant to penicillinase, but can be given with beta47 lactamase inhibitors to extend their microbial spectrum. Amox is more completely absorbed after oral administration and food interferes less with its absorption, therefore LESS diarrhea. c. Anti-pseudomonal penicillins (not resistant to penicillinase) i. Ticarcillin and Piperacillin: given parenterally, effective against Pseudomonas aeruginosa and enterococci and Bacterides fragilis. 4. Beta-lactamase inhibitors: Clavulanic Avid, Sulbactam, Tazobactam (combat penicillin resistance) a. Resemble beta-lactam molecules but have weak or no antibiotic activity. They act as potent, irreversible inhibitors of beta-lactamase. Beta-lactamase inhibitors extend antimicrobial spectrum of accompanying penicillin if bacterial resistance is due to beta-lactamase destruction AND if inhibitor is active against the particular beta-lactamase. b. Clavulanic Acid + amoxicillin and ticarcillin (oral). Sulbactam + ampicillin (parenteral). Taxobactam + piperacillin (parenteral). 48 1. Vancomycin a. Tricyclic glycopeptide acts by inhibiting cell wall synthesis at site different from penicillin (blocks linear polymerization, Stage 2) b. Poor oral absorption, administed IV (except for GI tract indications), excreted through kidneys (in renal failure half-life extended 6-10 days) c. Adverse reactions: chills, fever, rash (infusion related), ototoxicity is most SEVERE (pretreat with acetaminophen and diphenhydramine) d. Antimicrobial Spectrum/Clinical uses: use reserved from situations when less toxic agents are ineffective or not tolerate (Pen allergy) 2. Daptomycin a. Cyclic lipopeptide, parenteal, once dailym more rapidly bactericidal alternative to vancomycin. Mechanism involves action at bacterial membrane and loss of intracellular ions leading to cell death. Active against vancomycin resistant strains of staph (VRSA) and enterococci (VRE). Side effects: GI disorders, fever, HA, dizziness). 3. Cephalosporins a. Structure b. Relative to Pen G and V, cephalosporins have broader spectrum of action against gram – bacteria, less susceptibility to penicillinase, but cephalosporinases are emerging, less cross-reactivity in penicillin sensitive patients (1-5%), classified into 3 or 4 generations, breadth of activity against gram – bacteria is basis for classification. c. Absorption: acid-stable, orally given include: cephalexin, cephradine, cefadroxil (1st gen), cefaclor, cefuroxime (2nd gen), cefdinir, cefpodoxime, cefprozil (3rd). ALL OTHERS: IV/IM only. d. Distribution: penetrate tissues and fluids (including placenta) except CSF and brain. Major feature of 3rd gen CEPH is ability to penetrate into CSF. e. Metabolism-Excretion: primarily excreted through kidneys. Cefotaxime is the only to undergo metabolism. f. Classifications 49 i. First Generation: Cefaxolin, Cephradine, Cephalexin, effective against gram + cocci, gram – cocci and some gram – bacilli. Rarely drugs of first choice, antibacterial spectrum like amoxicillin, more stable than penicillins to many beta-lactams. Cefaxolin is prototype because low cost, low toxicity, good penetration into tissues an 90 min half-life (longer) ii. Second Generation: Cefuroxime, Ceftin, Cefotetan, Cefoxitin, Cefaclor, greater activity against gram – bacteria than 1st gen. Little to no activity against Pseudomonas. Increased spectrum of activity is due to increased penetration of gram – envelope and increased affinity for penicillinbinding proteins. Active against anaerobes like Bacteroides. iii. Third Generation: Cefdinir, Cefotaxime, Ceftraixone, Ceftazidime, Cefixime, Cefpodoxime, Cefprozil. Expanded gram – coverage compared to 2nd gen. More active against enteric gram – bacilli. Ceftazidime has moderate antipseudomonal activity. iv. Fourth Generation: Cefepime, similar to 3rd gen, but more resistant to chromosomal and extended spectrum beta-lactamases, good against Pseudomonas species g. Toxicity: well tolerated due to high selective toxicity i. Allergy/hypersensitivity: anaphylaxis, skin rash, nephritis, hemolytic anemia, rxns not as severe as with penicillins. Cross-hypersensitivity with penicillins (1-5%). Should not be given to pt with HX of immediate sensitivity to penicillin. ii. Other: nausea, vomiting, diarrhea. Super infection: with 2nd and 3rd gen (broader spectrum). Action to suppress intestinal flora can intensify effort of oral anticoagulants. h. Antimicrobial Spectrum/Clinical Uses 50 WEEK 4 Antimicrobials III Carbapenems: Imipenem-Cilastatin, Meropenem, Ertapenem Protein synthesis inhibitors Macrolides: Erythromycin, Clarithromycin, Azithromycin Tetracyclines: Tetracycline, Doxycycline, Minocycline Lincosamides: Clindamycin Synthetic: Chloramphenicol Aminoglycosides: Tobramycin, Gentamicin, Neomycin, Streptomycin Streptogramins: Quinupristin / Dalfopristin Oxazolidinones: Linezolid Miscellaneous Antibiotics Fluoroquinolones: Ciprofloxacin, Levofloxacin, Moxifloxacin Nitrofurantoin, Metronidazole Antimicrobial Resistance Mechanisms 1. Discuss the issues associated with an increased use of antimicrobial agents in the past several decades both in terms of human medicine and agriculture. a. World-wide problem, 90,000 people become ill with infections caused by MRSA and 15,000 of them die/year. b. Affects clinical outcome - associated with death rates c. Results in higher healthcare costs, Estimated >$5 billion, More expensive drugs d. Leads to prolonged hospitalization e. Increased challenges for appropriate management: Empiric therapy and Directed therapy 2. What are the major selective pressures that drive the emergence of antimicrobial resistance? a. Antibiotic use for growth promotion, prophylaxis and therapy (FOOD/ANIMALS) b. Antibiotic use for therapy and prophylaxis (HOSPITALIZED PATIENTS) 3. What are the major reservoirs of resistant organisms? a. Food/Animals, Humans in community, Hospitalized patients 4. What are the indicators that antimicrobial resistance is a global issue and not merely related to the increased usage of antibiotics in doctors’ offices and hospitals in developed countries? a. Substaintially more antibiotics are used in feedlots that in medicine. In some third-world countries, the antibiotic chloramphenicol is still used in animal feed lots to increase beef productivity. b. Resistant organisms can arise in a single location and be transmitted to other parts of the world. Even in seemingly isolated locations the identical resistance mechanisms exist that emerged only a few years earlier in distant countries. Human movement in today’s world makes the spread of resistance very easy and fast. 5. Describe the general mechanisms, for example intrinsic and acquired, associated with antimicrobial resistance. a. Intrinsic: Resistance that is already present in particular types of bacteria, Gram-negative bacteria are intrinsically resistant to some unmodified penicillins by the nature of their outer membrane 51 i. Biofilms: highly resistant structure which is composed of a matrix (usually polysaccharide) surrounding a group of bacteria. Bacteria in the biofilm are in a quiescent state of growth and they are highly resistant to antibiotics because the compounds require actively growing cells. Also, some antibiotics may not be able to diffuse through the matrix and would not reach a clinically useful concentration. ii. Efflux Pumps: mechanism for pumping out toxic substances from their cytoplasma through their cell membranes. A single efflux pump can pump out completely different classes of antibiotics. b. Acquired: Organisms acquire resistance from chromosomal mutations, or by the exchange of genetic material with resistant bacteria, “infectious antibiotic resistance” because is can be transmitted from one type of bacteria to another i. Resistance Transfer Factors (RTF): plasmids that carry genes for their own replication (oriR) and genes (tra) from their own transfer from 1 bacteria to another. Does not have to be of the same species or genera. RTF’s may or may not have antibiotic resistance genes. If a bacteria has a resistance factor plasmd (one that has genes for antimicrobial resistance)the RTF can mobilize that resistance factor to another cell wither by integrating the resistance factor into its DNA or it can provide transfer functions (sex pili) for the resistance factor that cannot transfer itself. ii. IS elements: DNA pieces that carry genes encoding proteins from their own transmission from 1 replicon (plasmid, bacteriophage, chromosome) to another. They cannot transmit themselves from 1 bacteria to another except by hopping into a piece of DNA that already has the ability to transfer itself. They cannot replicate autonomously. iii. Transposons: DNA fragments that are composed of 2 ID elements with extra baggage DNA (usually antibiotic resistance genes, or virulence factor genes or metabolic factors) between the IS elements. They can hop from 1 replicon to another and some can leave a copy of themselves behind when they leave (replicative transposition). Other cannot leave a copy behind. Cannot exist separately from another replicon (like IS elements) 1. Conjugative transposons: transposons that can mobilize themselves from 1 cell to another, they have acquired transfer genes like the RTF’s have, but still need another replicon for survival. 6. Discuss the several major biochemical and physiological mechanisms by which bacteria become resistant to antimicrobial agents. Describe how the alteration in the regulation of a gene can lead to resistance. Which of these mechanisms are responsible for clinically significant resistance? a. Mutational alteration of the target of the antibiotic: the resistant organisms would be less of a “fit” than the susceptible organisms because of altered target i. Examples: mutation in a ribosomal protein resistance to AG, mutation in cell wall structure beta-lactam resistance b. Enzymatic alteration of the antibiotic target i. Examples: vancomycin resistance, series of enzymes that alter the peptidoglycan precursor target of vancomycin. Erythromycin, methylation of antibiotic target, a ribosomal protein, the gene encoding this enzyme is regulated by erythromycin. c. Enzymatic alteration of the antibiotic: genes for this resistance usually are associated with mobile genetic elements (plasmid or transposon), but not always. i. Beta-lactams ii. AG’s modifying enzymes—all types leads to clinically significant resistance to AG’s d. Transport of the antibiotic out of the cell 52 i. Efflux pumps that transport different classes of antimicrobials OUT e. Alteration in the expression of endogenous genes that lead to antimicrobial resistance i. A mutation in promoter of gene encoding a porin that is required for antibiotic uptake ii. Mutation in promoter of inducible gene results in constitutive expression of enzyme that inactivates an antibiotic. f. Routing of metabolic pathway around the antimicrobial target i. Sulfonamides inhibit enzyme dihydrofolate reductaseinterference in folic acid and subsequent pyrimidine synthesis in bacteria. 7. What is meant by significant? “inducible resistance", and can it be clinically a. If an isolate of staphylococcus tests resistant to erythromycin and susceptible to clindamycin (a lincosamide), it could still possess the inducible (iMLSB) resistance mechanism. b. Macrolide-Lincosamide-Streptogramin B [MLSB] resistance is usually encoded by ermA or ermC genes. c. Induction tests to determine clindamycin resistance utilize erythromycin and clindamycin disks located on an agar plate in close proximity. d. This can be performed by the standard disk-diffusion procedure. After overnight incubation, if there is a flattening of the clindamycin zone adjacent to the erythromycin disk ("D-zone"); this indicates that the organism has iMLSB resistance. e. Organism is induced to turn on resistance genes to clindamycin in the presence of erythromycin (a strong inducer) Clinical Antibiotic Use: Bacterial Resistance and Antibiotic Resistance 1. Describe the laboratory tests used to determine measures of antimicrobial susceptibility. a. Tube Dilution (Broth Dilution) Susceptibility Test i. Serial dilutions of an antibiotic are made in liquid medium which is inoculated wth standardized # of organisms and incubated a certain time. The tubes are examined for bacterial growth (turbidity). The tube with the lowest concentration of antibiotic that has NO visible growth is the MIC. (in example, MIC is 3.1) 53 b. Disk diffusion or Kirby-Bauer Test i. Suspension of isolate at specific concentration is spread evenly onto agar in petri dish. Disks with defined concentrations of different antibiotics are placed onto agar surface. After incubation the diameter of a clear circular zone of inhibition of growth is measured. The diameter is proportional to the sensitivity of the organisms to the antibiotic. Using reference tables the size of the zone can be linked to the MIC and the organism can be classified as Susceptible, intermediate or resistant. c. E test i. Use a strip with a gradient of different concentrations of antibiotic. Strip is placed onto agar that has been prepared with isolate of interest. After incubation, an ellipse of inhibition is created and where the ellipse meets the strip, the MIC can be read. No tables are needed. 2. Define a. antimicrobial susceptibility: MIC is less than antibiotic levels (maximum dose) that can be achieved in the blood (exceptions: H. influenzae, S. pneumoniae) b. MIC: minimum inhibitory concentration is the lowest concentration of an antimicrobial that will inhibit the visible growth of a microorganism after overnight incubation. Variables for MIC: growth media, pH of growth media (pH can affect drug activity, antibiotic specific. Low pH ↓ aminoglycoside effect, pH effect are controlled in lab but in vivo pH is different), concentration of drug you need to inhibit bacteria (organism concentration, higher=higher drug concentration). c. MBC: minimum bactericidal concentration is the lowest concentration of the antibiotic that kiss 99.9% of the original inoculum in a given time. d. Bacteriostatic: inhibits bacterial growth, MBC=MIC e. Bactericidal: kills bacteria, MBC ˃˃˃MIC 3. Discuss the variables that affect clinical outcome of antimicrobial use a. HOST i. Organism concentration at site of infection, pH in the site of infection, antibiotic concentration and its penetration to the site of infection (urine= 100X % [plasma concentration of drug, soft 54 tissue=50-100%, lung= 50-100%, joint=50-100%, abscess= 5-10% and CSF= 5-10%), host defenses (immunocompromised hosts), some antibiotics depend on immune system to kill the bacteria (they just inhibit growth). 4. Discuss clinical and economic considerations for parenteral vs oral administration of antimicrobials a. CLINICAL i. Determine diagnosis, Consider Age and Preexisting Dx, Define common organsims, Determine likely resistance, Obtain proper cultures*, Initiate logical empiric therapy, Modify parenteral therapy based on cultures and patient response, Consider home IV or PO therapy (* Serious infections, immune compromised or poor response to prior therapy) ii. Switch to PO from IV: Define infection, Determine cause of infection & antimicrobial susceptibility, Achieve favorable response to IV, Determine if comparable levels can be achieved with PO doses, Assure compliance, Measure levels, Follow clinical response b. ECONOMIC i. 100 Pts, 6 wk treatment course with IV or PO after 5 hospital days, Hosp Day = $1000 IV Atb = $100/day, PO Atb = $10/day, Home Care = $300/day IVC complication rate 3/1000 pt days 5. Discuss the patterns and sources of the development of antimicrobial resistance a. Bacteria evolve as very fast rates and can develop mechanisms of resistance to antibiotics faster than we can develop new antibiotics. The more an antibiotic is used, the faster the resistance develops. b. Where antibiotics are used: human use= 50%, 20% of that is hospital and 80% is community (20-50% is unnecessary), agricultural use=50%, 20% of that is therapeutic and 80% is growth promotion and prophylactic (40-80% highly questionable). c. 55 6. Discuss the strategies to deal with emerging antimicrobial resistance 56