tpj12090-sup-0015-MethodsS1
advertisement

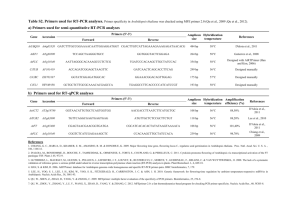
Supplemental methods: Cloning: pCPK28::GUS (pI2-pCPK28-GUS-mcs2): An S3 terminator was amplified by PCR from plasmid pPLEX3001 (Schünmann et al. 2003) using primers 401 and 402 and the ends of the resulting fragment were cut with XbaI and NotI. The 35S terminator of vector pam-PATMCS (accession number AY436765) was replaced by the S3 terminator using the vector’s XbaI and NotI restriction sites, generating the construct pI1. From genomic DNA of A. thaliana, ecotype Col-0, a promoter fragment of 1803 bp of AtCPK28 was amplified by PCR with primers 366 and 525. The promoter fragment was cut at the ends with AscI and EcoRI and used to replace the 35S promoter of pI1, released via AscI and EcoRI. This cloning step generated pI2-pCPK28. A multiple cloning site (mcs2) made from annealed primers 508 and 509 was cloned into pI2-pCPK28 opened with NotI and BstXI, generating pI2-pCPK28-mcs2. A β-Glucuronidase (GUS) gene was amplified with primers 403 and 438 from a GUS-vector introducing EcoRI and SmaI restriction sites at the ends. These sites were used to clone the GUS gene 3’ of the CPK28 promoter in pI2-pCPK28-mcs2 generating pI2-pCPK28-GUSmcs2. VK variants of CPK28 (pXCS-CPK28-VK-YFP and pXCS-CPK28-VK-D188A-YFP): The VK region of CPK28 was amplified with specific VK-primers from pXCSG-CPK28-YFP and pXCSG-CPK28-D188A-YFP and subcloned into pJET1.2 (Fermentas). Coding sequences were confirmed by sequencing and transferred to the binary expression vector pXCS–YFP (Feys et al. 2005) opened with HindIII and SmaI. pXCSG-pSUC2 and pXCSG-pKNAT1: From genomic DNA of A. thaliana, ecotype Col-0 promoter fragments of SUC2 and KNAT1 were amplified by PCR (An et al. 2004) (Primers see Supplemental Table S1). Promoter fragments were cut at the ends with AscI and HindIII and used to replace the 35S promoter of pXCSG-YFP, released via AscI and HindIII. This cloning step generated pXCSG-pSUC2-YFP or pXCSG-pKNAT1-YFP, respectively. Transient protein expression in Arabidopsis protoplasts: The preparation and transfection of Arabidopsis protoplasts was carried out as described (Wu et al. 2009). To exclude phosphorylation by endogenous CPK28, approximately 2 × 106 protoplasts derived from cpk28-1 plants were transfected with 100 μg DNA of each expression construct, harvested 16 – 18 h after transfection, and frozen in liquid nitrogen. For protein extraction, frozen pellets were resuspended by vortexing in extraction buffer [100 mM Tris-HCl (pH 8,0), 200 mM NaCl, 20 mM DTT, 10 mM NaF, 10 mM NaVO4, 10 mM β- glycerole-phosphate, 0,5 mM AEBSF, 100 μg/ml avidin and 1 × protease inhibitor mixture (Sigma)]. Crude extracts were centrifuged at 20,000 × g for 5 min at 4°C. The supernatant was transferred to fresh tubes and incubated with Strep-Tactin MacroPrep (IBA) for 30 min at 4 °C. Immobilised protein was washed five times with Wash buffer (100 mM Tris-HCl (pH 8,0), 200 mM NaCl, 2 mM DTT, 10 mM NaF, 10 mM NaVO 4, 10 mM β-glycerole-phosphate, 0,05 % TritonX-100) and, after acetylation of cysteins with iodacetamide, subjected to trypsin (Promega) digestion in 50 mM ammonium bicarbonate overnight at 37°C. Tryptic digest solutions were separated from the Streptactin matrix by centrifugation, transferred to new reaction tubes and vacuum-dried at 30°C. Mass spectrometry: Tryptic peptide mixtures were analysed by selected reaction monitoring using nanoflow HPLC (Proxeon Biosystems, www.proxeon.com) and a triple quadrupole mass spectrometer (TSQ Quantum Discovery Max, Thermo Scientific) as mass analyzer. Peptides were eluted from a 75 µm analytical column (Reprosil C18, Dr. Maisch, www.dr-maisch.com) on a linear gradient running from 10 % to 30 % acetonitrile in 50 minutes and were ionized by electrospray directly into the mass spectrometer. Specifically, phosphorylated and nonphosphorylated forms of the peptides FHDIVGSAYYVAPEVLK, ISLHEFR, and LTAAQALSHAWVR were analysed. Selected reaction monitoring (SRM) was used to quantify the abundance of phosphorylated and unphosphorylated forms of the respective peptides within each sample. The quadrupole Q1 was set as a mass filter for the parent ion, while Q3 was set to monitor specific fragment ions. Mass width for Q1 and Q3 was 0.7 Da, scan time was set to 5 milliseconds. Suitable fragment ions for SRM were selected experimentally determined and optimised using the software Pinpoint (Thermo Scientific). For each peptide, at least three fragment ions were used. Data analysis merging of fragment ion information to peptide average was also done using the Pinpoint software. For each phosphorylated and unphosphorylated peptide, intensities sums of three to five most intense fragment ions were displayed. Additional hormone treatments: Plants were grown on soil under long-day conditions (16 h light, 8 h dark) at 22°C unless otherwise stated. Hormone treatments were performed by spraying plants with either 0,95 % EtOH (mock), 100 µM Epibrassinolide (Sigma) or 100 µM IAA (Sigma) every other day. Plants were evaluated for the cpk28 phenotype 45 dag. Germination assay: Surface-sterilized seeds were plated on 0,5x Murashige and Skoog (MS) medium (pH 5.8) (Duchefa, www.duchefa.com) supplemented with 0.8% agar. To examine germination, seeds were generally first incubated for 2 d at 4°C and then grown at 22°C under LD conditions. Seed germination was quantified by scoring radicle emergence at the time points indicated in the figures. Presented are the averaged results of three independent seed batches (for each batch, WT and cpk28 mutant seeds were harvested at the same time, respectively) (n ≥ 36 per batch and genotype). Hypocotyl elongation assay: Surface-sterilized seeds were sown on 0,5x MS medium containing 0.8% agar supplemented with or without GA3 or PAC. Plates were incubated for 2 d at 4° C and then grown at 22°C in the dark or under LD conditions for 6 days. Hypocotyl lengths were measured with the ImageJ software. Protein extraction and Western Blot: Leaf material of transgenic lines was harvested and ground in liquid nitrogen, and proteins were extracted with extraction buffer (Franz et al. 2011). Proteins were separated on a 12% SDS/PAGE gel and analysed by Western blot. YFP-fusion protein detection was performed by anti-GFP (Roche, www.roche-applied-science.com) and an anti-mouse AP-conjugated antibody (Sigma, www.sigmaaldrich.com). Protein purification: Tagged CPK28 variants were transiently expressed in N. benthamiana as described previously (Witte et al. 2004). 0,5 g of N. benthamiana leaf material was ground in liquid nitrogen and resuspended in 1 ml extraction buffer (100 mM Tris/HCl pH 8.0, 5 mM EGTA, 5 mM EDTA, 100 mM NaCl, 20 mM DTT, 0.5 mM AEBSF, AL-Mix (2µg/ml antipain and 2µg/ml leupeptin), 2 µl/ml protease inhibitor cocktail (Sigma), 10 mM NaF, 10 mM Na3VO4, 10 mM -glycerolphosphate, 100 µg/ml avidin and 0.5% (v/v) Triton X-100). After removing insoluble debris, the supernatant was incubated with 40 µl Strep-tactin beads (IBA, www.ibalifesciences.com) and tagged proteins were immobilised by gentle end-over-end-rotation at 4°C for 30-60 min. Proteins were purified by washing 3-5 times with washing buffer (100 mM Tris pH 8,0, 150 mM NaCl, 1 mM EDTA, 2 mM DTT). Immobilised proteins were stored in washing buffer and transferred to buffer E (50 mM HEPES pH 7,4, 2 mM DTT, 0,1 mM EDTA) before the in vitro kinase assay. Kinase activity assay: Equal protein amounts used in kinase assays were determined by prior SDS-Gel electrophoresis and Western Blot analysis via Strep-Tactin-HRP or -AP conjugate (IBA). Protein purity was determined by detection of a single protein band on a Coomassie gel. Phosphorylation of Syntide 2 was assessed by the P81-filter-binding method as described (Romeis et al. 2001). The final reaction mixture (30 µl) contained 35 mM HEPES pH 7.4, 10 mM MgCl2, 1,4 mM DTT, 0.07 mM EDTA, 10 µM Syntide 2, 10 µM ATP, 3 µCi [γ-32P]ATP and different concentrations of CaCl2 or 2 mM EGTA as indicated. The reaction was carried out for 20 min at 30°C and was stopped by adding 3 µl of 10% phosphoric acid. Subsequently, 20 µl of each reaction was dropped on P81 paper and dried at room temperature. Three washing steps with 1% phosphoric acid removed free [γ-32P]-ATP and incorporated 32P was determined by liquid scintillation counting. An, H., Roussot, C., Suarez-Lopez, P., Corbesier, L., Vincent, C., Pineiro, M., Hepworth, S., Mouradov, A., Justin, S., Turnbull, C. and Coupland, G. (2004) CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Development, 131, 3615-3626. Feys, B.J., Wiermer, M., Bhat, R.A., Moisan, L.J., Medina-Escobar, N., Neu, C., Cabral, A. and Parker, J.E. (2005) Arabidopsis SENESCENCE-ASSOCIATED GENE101 stabilizes and signals within an ENHANCED DISEASE SUSCEPTIBILITY1 complex in plant innate immunity. Plant Cell, 17, 2601-2613. Franz, S., Ehlert, B., Liese, A., Kurth, J., Cazale, A.C. and Romeis, T. (2011) Calciumdependent protein kinase CPK21 functions in abiotic stress response in Arabidopsis thaliana. Mol Plant, 4, 83-96. Romeis, T., Ludwig, A.A., Martin, R. and Jones, J.D.G. (2001) Calcium-dependent protein kinases play an essential role in a plant defence response. EMBO J, 20, 5556-5567. Schünmann, P.H.D., Llewellyn, D.J., Surin, B., Boevink, P., Feyter, R.C.D. and Waterhouse, P.M. (2003) A suite of novel promoters and terminators for plant biotechnology. Functional Plant Biology, 30, 443-452. Witte, C.P., Noel, L.D., Gielbert, J., Parker, J.E. and Romeis, T. (2004) Rapid one-step protein purification from plant material using the eight-amino acid StrepII epitope. Plant Mol Biol, 55, 135-147. Wu, F.H., Shen, S.C., Lee, L.Y., Lee, S.H., Chan, M.T. and Lin, C.S. (2009) TapeArabidopsis Sandwich - a simpler Arabidopsis protoplast isolation method. Plant Methods, 5.