Doc S1 of Supplementary Material
advertisement

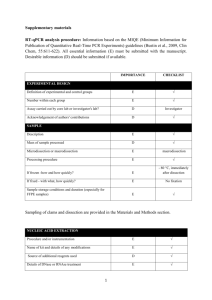
Supplementary Material to Rouvinen-Watt et al., page 1 Supplementary Material to Evidence of endoplasmic reticulum stress and liver inflammation in the American mink Neovison vison with benign hepatic steatosis by Rouvinen-Watt et al. CONTENT Doc S1 of Supplementary Material…………………………………………….……………….1 Author contributions ..................................................................................................................... 1 RNA extraction ............................................................................................................................. 1 Reverse transcription .................................................................................................................... 1 qPCR protocol .............................................................................................................................. 2 Validation ..................................................................................................................................... 2 Data analysis ................................................................................................................................. 2 MIQE checklist ............................................................................Error! Bookmark not defined. Table S1 of Supplementary Material…………………………………………….……………..7 Table S2 of Supplementary Material…………………………………………….……………..7 Table S3 of Supplementary Material…………………………………………….……………..8 References to Supplementary Material…………………………………………….…………..8 Author contributions K. Rouvinen-Watt, P. Nieminen, and A.-M. Mustonen planned this research and KRW oversaw all experimental procedures. C. Pal, T. Martin, L. Harris, D. Kryzskaya, and V. Kärjä carried out the laboratory analyses. Data analysis was performed by KRW, T. Astatkie, CP, TM, and PN. R. Tammi and M. Tammi contributed to the interpretations of the data. Manuscript writing was led by KRW, with contributions from all co-authors. RNA extraction Mink livers were obtained (by macrodissection) within 15–25 min from time of death. Samples were immediately snap frozen in liquid nitrogen and kept at –85°C until RNA extraction, six months later. Total RNA content was extracted from approximately 30 mg tissue, with homogenization using the Kimble® Kontes Pellet Pestle and QIAShredder homogenizers. RNA was treated with RNase free DNase I (Qiagen, Toronto, ON, Canada) using the manufacturer’s on-column treatment protocol. There was an average yield of 2.28 ± 0.06 µg RNA/mg tissue. Sample purity was assessed by an A260:A280 ratio of 1.8–2.1. Reverse transcription Total RNA (1.0 µg) was reverse transcribed with 2.5 U/µL MultiScribe™ Reverse Transcriptase in a reaction volume of 20 µL at 37°C for 120 min, using the High Capacity cDNA Reverse Transcription Kit with RNase inhibitor, catalog# 4374966 (Applied Biosystems, Carlsbad, CA, USA). 0.1X and 0.01X dilutions of cDNA were prepared in 10 mM Tris-Cl, pH=7.6, and stored Supplementary Material to Rouvinen-Watt et al., page 2 at –30°C. GAPDH qPCR run on a set of no reverse transcription controls showed no genomic contamination in 42 samples, minor contamination (Cq=31.2) in one sample, and trace contamination in the remaining 17 samples (Cq>35). qPCR protocol Primers, purified by reverse-phase cartridge separation, were purchased from Sigma (SigmaAldrich Canada Co, Oakville, ON, Canada). Assays were run in a reaction volume of 10 µL, using 1 µL of 0.1X diluted cDNA, using 384-well white plates, catalog #04729749001 (Roche Diagnotics Canada, Laval, QC, Canada). Each sample was run with three technical replicates at the qPCR stage, and each treatment group included five technical replicates. All assays except GRP78 were run with 1X GoTaq® Flexi buffer, 2.5 mM MgCl2, 0.2 mM dNTP (each), 0.8 U GoTaq® HotStart Polymerase (Promega, Fitchburg, WI, USA), and 1X EvaGreen® (Biotium Inc, Hayward, CA, USA). The GRP78 assay was run using 1X SsoFastTM EvaGreen® mastermix (Bio-Rad Laboratories Inc., Hercules, CA, USA), 500 nM primers, with a hot start at 95°C for 2 min followed by 35 cycles of denaturation at 95°C for 5 s, combined annealing and extension at 58°C for 20 s. Validation The specificity of each qPCR assay was confirmed by verification of the size of the amplicon on agarose gel electrophoresis (gel photographs available on request), and the presence of a single peak in a melt curve. No amplification was seen in the no template controls (n=6). The limit of detection for each assay was at or below the lower limit of the linear dynamic range as reported in Table S3, as shown by a linear standard curve to this point. To test for inhibition, qPCR results of standards spiked with pooled cDNA (0.1X dilutions) were compared with those of pooled cDNA (0.1X dilution) alone, and standards run separately. No inhibition was seen. Data analysis Lightcycler® 480 Software, release 1.5.0.39 (Roche, Indianapolis, IN, USA) was used to obtain quantification cycles (Cq) results for each sample, employing the “second derivative maximum” method. Each qPCR run included a standard curve (a 5-fold dilution series from a known amount of target cDNA, created from synthetic copy RNA), which was used to calculate the expression of the target gene in each sample. Each run was examined for anomalous amplification curves, and all were within acceptable norms, so that no outliers were removed. To normalize each sample, the geometric mean of the replicate concentrations of a target gene was divided by the geometric mean of the replicate concentrations of the reference gene. Supplementary Material to Rouvinen-Watt et al., page 3 MIQE Checklist according to Bustin et al. (2009) ITEM TO CHECK IMPORTANCE CHECKLIST WHERE IN THE MANUSCRIPT Definition of experimental and control groups E Yes Methods and materials: Animal experiment Number within each group E Yes Methods and materials: Animal experiment Assay carried out by core lab or investigator's lab? D Yes All experiments carried out in investigator’s lab. Acknowledgement of authors' contributions D Yes S1: Author contributions E Yes Methods and materials: Quantitative real-time polymerase chain reaction Volume/mass of sample processed D Yes S1: RNA extraction Microdissection or macrodissection E Yes S1: RNA extraction E Yes S1: RNA extraction If frozen - how and how quickly? E Yes S1: RNA extraction If fixed - with what, how quickly? E Yes S1: RNA extraction E Yes S1: RNA extraction EXPERIMENTAL DESIGN SAMPLE Description Processing procedure Sample storage conditions and duration (especially for FFPE samples) NUCLEIC ACID EXTRACTION Procedure and/or instrumentation Methods and materials: Quantitative real-time polymerase chain reaction Methods and materials: Quantitative real-time polymerase chain reaction E Yes Name of kit and details of any modifications E Yes Source of additional reagents used D n/a Details of DNase or RNase treatment E Yes S1: Validation Contamination assessment (DNA or RNA) E Yes Nucleic acid quantification E Yes Instrument and method E Yes S1: Reverse transcription Methods and materials: Quantitative real-time polymerase chain reaction Methods and materials: Quantitative real-time polymerase chain reaction Purity (A260/A280) D Yes S1: RNA extraction Yield D Yes S1: RNA extraction E Yes Methods and materials: Quantitative real-time RNA integrity method/instrument Supplementary Material to Rouvinen-Watt et al., page 4 polymerase chain reaction RIN/RQI or Cq of 3' and 5' transcripts E ND Electrophoresis traces D Yes Available on request E Yes S1: RNA extraction E Yes Methods and materials: Quantitative real-time polymerase chain reaction and S1 Amount of RNA and reaction volume E Yes S1: Reverse transcription Priming oligonucleotide (if using GSP) and concentration E N/A Inhibition testing (Cq dilutions, spike or other) REVERSE TRANSCRIPTION Complete reaction conditions E Yes Methods and materials: Quantitative real-time polymerase chain reaction and S1: Reverse transcription Temperature and time E Yes S1: Reverse transcription Manufacturer of reagents and catalogue numbers D Yes S1: Reverse transcription Cqs with and without RT D* Yes S1: Reverse transcription Storage conditions of cDNA D Yes S1: Reverse transcription If multiplex, efficiency and LOD of each assay. E N/A Sequence accession number E Yes Table 1 Location of amplicon D Yes S1: Table S1 Amplicon length E Yes Table 1 In silico specificity screen (BLAST, etc) E Yes Primer BLAST run for all targets (Ye et al. 2012) Pseudogenes, retropseudogenes or other homologs? D No Information unavailable for mink. D No D Yes All amplicons analyzed using Mfold (Zuker 2003) E Yes S1: Table S1 E No Splice variation information unavailable for mink. Primer sequences E Yes Table 1 RTPrimerDB Identification Number D N/A Reverse transcriptase and concentration qPCR TARGET INFORMATION Sequence alignment Secondary structure analysis of amplicon Location of each primer by exon or intron (if applicable) What splice variants are targeted? qPCR OLIGONUCLEOTIDES Supplementary Material to Rouvinen-Watt et al., page 5 Probe sequences D** N/A Location and identity of any modifications E Yes None Manufacturer of oligonucleotides D Yes S1: qPCR protocol Purification method D Yes S1: qPCR protocol E Yes Methods and materials: Quantitative real-time polymerase chain reaction Reaction volume and amount of cDNA/DNA E Yes S1: qPCR Protocol Primer, (probe), Mg++ and dNTP concentrations E Yes S1: Table S2 and qPCR protocol Polymerase identity and concentration E Yes S1: qPCR protocol Buffer/kit identity and manufacturer E Yes S1: qPCR protocol Exact chemical constitution of the buffer D No Not available from manufacturer. Additives (SYBR Green I, DMSO, etc.) E Yes Manufacturer of plates/tubes and catalog number D Yes Complete thermocycling parameters E Yes Reaction setup (manual/robotic) D Yes Manufacturer of qPCR instrument E Yes S1: qPCR protocol Methods and materials: Quantitative real-time polymerase chain reaction and S1: qPCR protocol Methods and materials: Quantitative real-time polymerase chain reaction, S1: Table S2, and S1: qPCR protocol Methods and materials: Quantitative Real-Time Polymerase Chain Reaction Methods and materials: Quantitative real-time polymerase chain reaction Evidence of optimization (from gradients) D Yes Available on request Specificity (gel, sequence, melt, or digest) E Yes S1: Validation For SYBR Green I, Cq of the NTC E Yes S1: Validation Standard curves with slope and y-intercept E Yes S1: Table S3 PCR efficiency calculated from slope E Yes S1: Table S3 Confidence interval for PCR efficiency or standard error D No E Yes S1: Table S3 E Yes S1: Table S3 qPCR PROTOCOL Complete reaction conditions qPCR VALIDATION 2 r of standard curve Linear dynamic range Supplementary Material to Rouvinen-Watt et al., page 6 Cq variation at lower limit E Yes S1: Table S3 Confidence intervals throughout range D No Evidence for limit of detection E Yes If multiplex, efficiency and LOD of each assay. E N/A E Yes S1: Data analysis Cq method determination E Yes S1: Data analysis Outlier identification and disposition E Yes S1: Data analysis Results of NTCs E Yes Justification of number and choice of reference genes E Yes Description of normalisation method E Yes Number and concordance of biological replicates D Yes S1: Validation Methods and materials: Quantitative real-time polymerase chain reaction Methods and materials: Quantitative real-time polymerase chain reaction and S1: Data analysis Methods and materials: Animal experiment and S1: qPCR protocol Number and stage (RT or qPCR) of technical replicates E Yes S1: qPCR protocol Repeatability (intra-assay variation) E Yes S1: Table S3 Reproducibility (inter-assay variation, %CV) D No Power analysis D No Statistical methods for result significance E Yes Methods and materials: Statistical analysis Software (source, version) E Yes Methods and materials: Statistical analysis Cq or raw data submission using RDML D No S1: Validation DATA ANALYSIS qPCR analysis program (source, version) Supplementary Material to Rouvinen-Watt et al., page 7 Table S1 Target information for qPCR primer pairs. Sequence Location of Amplicon Location of forward Location of reverse accession number amplicon length primer (human exon) primer (human exon) GAPDH AF076283 36-116 84 7 7 GRP78 HQ003898 501-611 111 8 8 LPL AJ223493 1813-1908 96 10 10 MCP-1 HQ163895 149-266 118 2 3 TNF-a GU327784 81-205 125 3 4 Target gene Table S2 Thermocycling parameters for qPCR assays. Concentration primers (nM) 400 Number of cycles 40 Annealing temperature (°C) 60 Denaturation time (s) 10 Annealing time (s) 20 Extension time (s) 30 LPL 400 45 60 10 20 30 MCP-1 400 45 60 30 30 30 TNF-a 400 45 62 30 30 30 Target gene GAPDH Supplementary Material to Rouvinen-Watt et al., page 8 Table S3 qPCR validation for qPCR assays. Target Standard curve Linear dynamic Cq variation at Repeatability (intra-assay range (copies) lower limit variation) (CV of Cq, %) 103-108 0.07 0.10 Y-intercept Efficiency (%) r2 41.69 93.8 0.9999 –3.542 42.28 91.6 0.9990 103-108 0.00 0.25 LPL –3.513 43.29 92.6 0.9988 102-108 0.82 0.19 MCP-1 –3.465 47.41 94.4 0.9968 103-108 0.43 0.46 TNF-a –3.455 40.05 94.7 0.9968 25-108 0.06 1.11 gene Slope GAPDH –3.481 GRP78 References to Supplementary Material Bustin SA, Benes V, Garson JA et al (2009) The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin Chem 55:611–622 Ye J, Coulouris G, Zaretskaya I et al (2012) Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134 Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415