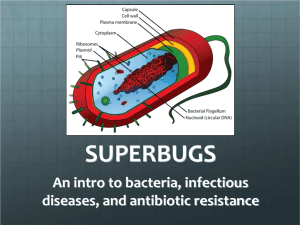
View/Open - Lirias
advertisement

Running title: Renaissance in antibiotic discovery RENAISSANCE IN ANTIBIOTIC DISCOVERY: SOME NOVEL APPROACHES FOR FINDING DRUGS TO TREAT BAD BUGS Bharat Gadakh and *Arthur Van Aerschot KU Leuven – University of Leuven, Department of Pharmaceutical and Pharmacological Sciences, Medicinal Chemistry, Rega Institute for Medical Research, 3000 Leuven, Belgium * Corresponding author: Prof. Dr. Arthur Van Aerschot Address: KU Leuven – University of Leuven, Medicinal Chemistry, Rega Institute for Medical Research, Minderbroedersstraat 10, 3000 Leuven, Belgium Direct Phone (32) 16 372624; Fax: (32) 16 337340; E-mail: Arthur.VanAerschot@rega.kuleuven.be Bharat Gadakh Address: KU Leuven – University of Leuven, Medicinal Chemistry, Rega Institute for Medical Research, Minderbroedersstraat 10, 3000 Leuven, Belgium Direct Phone (32) 16 337381; Fax: (32) 16 337340; E-mail: bharat.gadakh@rega.kuleuven.be 1 Abstract With the alarming resistance to currently used antibiotics, there is a serious worldwide threat to public health. Therefore, there is an urgent need to search for new antibiotics or new cellular targets which are essential for survival of the pathogens. However, during the past 50 years, only two new classes of antibiotics (oxazolidinone and lipopeptides) have reached the clinic. This suggests that the success rate in discovering new/novel antibiotics using conventional approaches is limited and that we must reconsider our antibiotic discovery approaches. While many new strategies are being pursued lately, this review primarily focuses only on a few of these novel/new approaches for antibiotic discovery. These include structure-based drug design (SBDD), the genomic approach, anti-virulence strategy, targeting non-multiplying bacteria and the use of bacteriophages. In general, recent advancements in nuclear magnetic resonance, X-crystallography, and genomic evolution have significant impact on antibacterial drug research. This review therefore aims to discuss recent strategies in searching new antibacterial agents making use of these technical novelties, their advantages, disadvantages and limitations. Keywords: acylated homoserine lactone, antibiotics, antibiotic discovery, antivirulence strategy, autoinducers, bacteriophages, genomic approach, non-multiplying bacteria, phage therapy, quorum sensing, resistance, screening strategies, structure-based drug design. 2 1. INTRODUCTION For more than seven decades, antibiotics are essential components of modern medicine and are one of the leading causes for increased life expectancy [1, 2]. However, over the last two decades, development of resistance to antibiotics poses a serious threat to public health worldwide [3, 4]. Nowadays, most of the lethal infections are caused by resistant pathogens like methicillin resistant Staphylococcus aureus (MRSA), vancomycin resistant Enterococcus (VRE), extended-spectrum lactamase producing enterobacteriaceae, Pseudomonas aeruginosa and Acinetobacter baumannii. Looking back at the timeline of antibiotic development from 1935 till present, it can be seen that the majority of the antibiotics in clinical use have been discovered during the 1940s-1960s and this period therefore is called the golden period of antibiotic discovery. In the past 50 years only two new classes of antibiotics (i.e. oxazolidinone and lipopeptides) have reached the clinic. With the emergence of multidrug resistant pathogens and the withdrawal of pharmaceutical companies from the infectious disease area, we are in danger that these resistant pathogens may send us back into the pre-antibiotic era leaving us without any protection against these resistant pathogens. So, we need to ask ourselves “where did we go wrong?” The factors contributing to this situation are numerous and complex [5-8]. These factors include the shifting research priorities of the pharmaceutical companies, the raised utility bar by Food and Drug Administration (FDA), limited market due to improved hygiene, regulatory hurdles, rapid development of resistance, etc. In addition, success in discovering antibiotics using conventional approaches like high throughput screening (HTS) and combinatorial chemical libraries is rare [1, 9, 10]. In order to fight these bad bugs (resistant pathogens), new classes of antibiotics must enter the clinic at regular intervals. Therefore, with alarming bacterial resistance and low productivity of conventional whole cell-based screening assays, we must consider alternative approaches for antibiotic discovery. However, also commercial considerations have driven pharmaceutical companies out of the antibiotic development area. Most infections can still be treated with current antibiotics at virtually no cost. With these at hand, companies hardly can expect therefore to get the same price for a new antibiotic as is now been paid for an antiviral or an anticancer drug. An extravagant example of pricing herein is seen with the antiviral drug sofosbuvir (SovaldiTM, Gilead Sciences), a once-daily oral nucleotide analog polymerase inhibitor for the treatment of chronic hepatitis C infection, where a 1000 $ per pill is charged, resulting in a total 85,000 $ treatment regimen Such examples of exorbitant costs in our opinion could also provoke a backlash against pharmaceutical industry by both government officials and private insurance companies [11]. In contrast, in view of the dwelling pipeline and the alarming rise in antimicrobial resistance, European Commission launched an action plan resulting in the New Drugs for Bad Bugs (ND4BB) initiative. The latter aims to join forces between public and private partners in order to bring new antimicrobials closer to patients and to boost research on antibiotics especially against Gram-negative 3 bacteria. Likewise, being aware of the lack of adequate reward for companies developing antibiotics, American government decided to stimulate antibiotic research via altered legislation. Hereto, the GAIN initiative (Generating Antibiotic Incentives Now), part of the FDA Safety and Innovation Act was signed into law in July 2012, which for infectious disease products provides a more speedy review and, if approved, provides companies with five additional years of market exclusivity to recoup their investments [12]. These initiatives have certainly boosted research on antibiotics lately, both in academia and in smaller biotech companies. This review aims to illustrate some recent approaches for discovery of new antibiotics and the advantages, disadvantages and experimental challenges facing their development. By no means is it meant to be exhaustive with many new ideas being pursued over the last years. All novel approaches can be classified into several strategies such as structure-based drug design (SBDD), the (post-)genomic approach, anti-virulence strategy, the use of bacteriophages, non-multiplying bacteria as a target, noncultivable bacteria as antibiotic sources, antibiotics from marine sources, the search for unexploited and vital targets, targeting resistance mechanisms (e.g. β-lactamase inhibitors), the RNAi approach, inhibition of peptidoglycan biosynthesis, methods to enhance uptake of inhibitory products, antimicrobial peptides, etc. However, in view of length considerations this review focuses on only the first five approaches mentioned above and the lessons they taught us. All these strategies, however, showed some successes and failures in their preliminary screening. Other approaches are reviewed elsewhere, e.g. the RNAi approach [13] or antimicrobial peptides [14, 15] and we refer to these. Many alternative therapeutic strategies are currently under consideration as well and cannot all be covered here. Industrial views on the topic can be found amongst others in the opinion papers of David Payne discussing the (industrial) challenges of antibacterial discovery. Innovation herein can take many forms, from new physicochemical properties for analogues of an existing class, over modifications to scaffolds overcoming resistance problems, to agents with new mechanisms of action, development of novel chemical classes or uncovering of new targets [16, 17]. Another recent review which could be consulted discusses the different platforms for antibiotic discovery [18]. In our last part, a brief discussion on bacteriophage therapy is included as it constitutes an often forgotten strategy to combat specific infections like in patients with serious burn wounds. However, the lack of a legal framework seriously hampers the use of this latter technique. 2. CLASSICAL OR CONVENTIONAL APPROACH Before moving on to a discussion on the new strategies, it is important to briefly look at the classical approaches of antibiotic discovery. One such approach involves the random screening of potential inhibitors (obtained from natural product or synthetic chemical libraries) in a whole cell assay, with growth inhibition or the death of a pathogen as an end point [9, 10]. The active compounds from 4 these screening activities are then tested in an in vivo model to determine efficacy and safety. Most of the clinically used antibiotics have been discovered using this approach. The ensemble of these antibiotics targets only a few essential cellular pathways such as cell wall biosynthesis, nucleic acid metabolism, protein synthesis and fatty acid biosynthesis. The conventional screening process gives valuable information on potency (minimum inhibitory concentration - MIC), the spectrum of activity and the ability of the compound to enter bacterial cells. However, the drawback of this approach is that it may eliminate potentially important compounds at the early stages of drug discovery as it is mainly based on two properties of the compounds: antibacterial activity and the ability to enter the bacterial cells. Is it however appropriate to demand both afore mentioned properties in a new compound at early stage? In retrospect, it might be better to get target specificity first and to optimize a lead to address other factors such as cell permeability. As this conventional strategy targets essential biosynthetic pathways in multiplying bacteria, these antibacterial agents are inactive or only partially active against non-multiplying bacteria [19, 20]. Following the ‘golden period’ of antibiotic discovery, subsequent improvements in the infectious disease area were achieved by chemical modifications of existing antibiotics. This approach is called the conservative approach which yielded short-term success. For example ampicillin and methicillin are derived from penicillin with an expanded spectrum and with increased resistance to β-lactamases respectively. Further, screening of vast chemical libraries was accelerated by high throughput screening (HTS). However, the hits identified from HTS often fail to reach the clinic due to poor selectivity, toxicity, inefficient uptake, narrow spectrum of activity etc. Therefore at this point of time, we need to revise our approaches for antibiotic discovery. 3. NEW/NOVEL APPROACHES FOR DISCOVERY OF ANTIBIOTICS Although rational design of antibiotics is a valid approach, it has not been practiced much due to the technical difficulties and limited understanding of the targets. However, recent technical advancements in nuclear magnetic resonance (NMR), X-ray crystallography, computational tools, biochemical and genetic tools have contributed significantly in antibacterial drug development. These new approaches of antibiotic discovery explore either novel cellular targets which are essential for survival of a pathogen or new antibacterial agents acting by a novel mode of action. As mentioned before, only the topics SBDD, the (post-)genomic approach, anti-virulence strategy, targeting nonmultiplying bacteria and the use of bacteriophages as biological antibacterial agents will be discussed in the next sections. 3.1. Structure-Based Drug Design (SBDD) 5 Drug discovery and development is a complex and multivariate process driven by economic and regulatory factors. In 1990s, the application of combinatorial chemistry along with HTS in the pharmaceutical industry significantly accelerated screening of vast chemical libraries against a potential target. However, there has not been a proportional increment in the number of new chemical entities (NCEs) reaching the approval. During the same time many pharmaceutical companies also pursued a target-based drug discovery approach for antibiotic discovery. This approach was mainly based on a combined application of the knowledge of the bacterial genome, HTS and SBDD. The technical advances in X-ray crystallography, NMR and computational tools and the possibility of crystallizing many vital bacterial targets contributed significantly to the use of SBDD in antibacterial drug discovery. Numerous literature reports contain a variety of examples where the SBDD approach has been used for hit identification and lead optimization of known antibiotics [16, 21-23]. Moreover, a better understanding of the molecular basis of resistance also opens up many opportunities where SBDD proved to be a powerful tool to fight resistant pathogens. Here, the development of a novel dihydrofolate reductase (DHFR) inhibitor, iclaprim has been described as an illustrative example where SBDD plays an important role to address the issue of bacterial resistance. Two very recent examples are added confirming the power of this matured technique. 3.1.1. SBDD: addressing the issue of resistance Dihydrofolate reductase (DHFR) is an essential bacterial enzyme which is responsible for synthesis of tetrahydrofolate from dihydrofolate. It has been used as an antibacterial target over the last five decades. Trimethoprim (TMP) (compound 1, Fig. 1) is a potent and selective inhibitor of DHFR (with Ki of 0.9 nM for Staphylococcus aureus DHFR vs Ki of 19 M for human DHFR) and is used in a synergistic combination with sulfamethoxazole (SMX) (compound 2, Fig. 1). The synergistic combination of TMP and SMX is marketed by Roche Pharmaceuticals as Bactrim™. After a long-term clinical use, not surprisingly, the effectiveness of these drugs is reduced due to the emergence of resistance. The extent of resistance varies among bacterial species and the geographical location. For example, in 2002, in the USA approximately 20% and in Korea 70% of S. aureus strains were found to be Bactrim™ resistant. The crystal structure of DHFR from the resistant stain suggests that resistance to TMP has occurred via a point mutation in the TMP binding site by substituting phenylanine at position 98 with tyrosine (F98Y). This mutation results in the loss of a hydrogen bond between the 4-amino moiety of TMP and the carbonyl oxygen of Leu6 [24-26]. Therefore, researchers at Roche made several attempts to eliminate the 4-amino group or to replace the 2,4-diaminopyrimidine core with other heterocycles. However, such replacement resulted in reduced potency as compared to TMP. Finally, combining detailed structural knowledge and the SBDD approach, they designed modified diaminopyrimidines 6 that would compensate for the loss of hydrogen bonding. A few key DHFR inhibitors have been depicted in Fig. 1 (compounds 3-5). Iclaprim (compound 3, Fig. 1) is the most studied compound in this series and adds favorable hydrophobic interactions via the iclaprim cyclopropyl group with Ile50 and Leu54 of mutant DHFR [27-29]. Iclaprim has better potency against both the wild type (15 fold) and the F99Y mutant (100 fold) strains as compared to TMP. From a properties standpoint, the water solubility of iclaprim is reduced by 20-fold as compared to TMP. Pharmacokinetic studies indicate that the human oral bioavailability of iclaprim is only 40% as compared to >90% for TMP (smaller and more water soluble). Recently, iclaprim was evaluated in a phase III clinical trial for treatment of patients with complicated soft structure skin infections (cSSSI). The clinical data demonstrated that iclaprim was safe and efficacious. However, additional clinical data were required by FDA to demonstrate efficacy in order to gain approval [29]. In 2010, Acino Pharma acquired the rights for this intravenous (i.v.) antibiotic efficacious against MRSA strains resistant to many antibiotics and beginning of 2014 was taken over by Pharma Strategy partners. The actual fate of iclaprim is unclear though. In addition and in brief, a more recent example with defensin mimetics targeting Lipid II highlights once more the power of SBDD. Lipid II, being an essential precursor in cell wall biosynthesis and having a high turnover rate has been established before as an ideal molecular target for development of antibiotics and several peptide antibiotics among which the glycopeptides vancomycin and teicoplanin have been described to target Lipid II [30]. Following their report on the functional interaction of the human defensin peptide HNP1 with Lipid II [31], the authors were able to cocrystallize and solve the structure of a HNP-1/Lipid II complex. Following establishment of pharmacophore models with the required orientation of side chains in space and in silico searching within libraries of drug-like compounds, the above uncovered detailed interaction allowed to identify low molecular weight compounds with strong similarity to the defensin HNP1. Several of these showed strong antibacterial activity and specific killing of S. aureus. The lead compound coined BAS00127538 (Compound 6, Fig. 1) herein most strongly bound to Lipid II, as shown by surface plasmon resonance. Inhibition of cell wall synthesis was proved to be the primary target, with membrane perturbation as a secondary mechanism [32]. The compound likewise was shown to strongly inhibit Acinetobacter baumanii further highlighting that small molecule inhibitors of Lipid II have the potential to be developed into broad-spectrum therapeutics [33]. A further recent example from Trius Therapeutics used the well-known sulfamate inhibitor Threonine-AMS as a starting point and came up with a series of potent and bacteria-selective threonyltRNA synthetase (ThrRS) inhibitors (Compound 7, Fig. 1) in which both the sugar and natural nucleoside base part were substituted for by heterocycles. The study relied on computational modelling using the structural information of the Thr-AMS/ThrRS complex. Nice antimicrobial activities were noted for the lead compounds against Haemophilus influenzae, efflux-deficient mutants of Escherichia 7 coli and against Burkholderia thailandensis [34]. All of these clearly establish the power of SBDD provided the necessary high resolution structural information is available. 3.1.2. Lessons learned from SBDD SBDD is a promising strategy for antibiotic discovery and in addition has been used for many other targets such as the DNA gyrase subunit B (GyrB) [35, 36], LpxC (a bacterial deacetylase essential for lipopolysaccharide biosynthesis) [37], methionyl-tRNA synthetase (MetRS) [38], -lactamase [39] and bacterial ribosome [40]. SBDD has proven its value for optimization of the lead structure and for addressing the issue of resistance. As compared to other strategies, SBDD fairly rapidly leads to discovery of compounds with antibacterial activities. In addition optimization of hits from HTS has been relatively effective. Although SBDD can be used to tailor the spectrum, activity of the compounds against multiple enzyme orthologs are often ignored, not reported or probably not measured. In addition, the choice of the target plays a vital role in determining the potency and spectrum of the new antibacterial agents. It is difficult to predict the degree of success at early stage because the lack of in vivo antibacterial activity despite nice in vitro activity may be due to other factors such as poor bacterial cell wall penetration. The SBDD strategy often affects drug properties such as hydrophilicity, lipophilicity and solubility, which in turn influence oral absorption and bioavailability (see also the issue iclaprim vs TMP). A comment should be added in the sense that in using SBDD we only discussed here the targeting of classical resistance elements. In addition, the “intrinsic resistome” as recently defined comprises all elements that contribute directly or indirectly to bacterial resistance to antibiotics, independent of horizontal gene transfer. Inhibition of these elements or genes will turn bacteria either more susceptible or more resistant to antibiotics dependent on the nature of the element, being for instance a plasmid-encoded -lactamase, an efflux pump or a transcriptional repressor of an efflux pump. The study of the intrinsic resistome indicates diverse possibilities of modulating antibiotic efficacy and is a research topic on its own, which has been very nicely covered recently by Martinez et al. [41]. 3.2 Genomic Approach In 1995, the complete bacterial genomic sequence of H. influenzae was published, which was considered as the beginning of the ‘genomic era’ [42]. Subsequently, the complete genomic sequence of Saccharomyces cerevisae (1996) [43] and E. coli K-12 (1997) [44] were published. In recent years, there has been an explosion in the amount of available genome data which has had a great impact on antibacterial drug discovery [45, 46]. In fact more than 1000 complete bacterial genome sequences are 8 available for comparative analysis in antibiotic drug discovery and many more are currently in progress. It was assumed that the bacterial genome would reveal a treasure of unknown and therefore unexploited targets for antimicrobial therapy and would speed up the process of drug discovery. In retrospect, this approach yielded limited success owing to difficulties in the target selection, the screening procedures and the compound pools used as source of inhibitors. The process of antibiotic discovery using a genomic approach is represented schematically in Fig. 2A. As a bioinformatics-driven approach the target selection is limited to the areas where a full package of information related to a particular gene is known. But it does provide a framework for comparative analysis of genes and prioritization of new targets which can be used to investigate genes of unknown function. Many pharmaceutical companies have embarked on programs to find antibacterial agents using a genomic approach. Illustrative examples include large screening campaigns of GlaxoSmithKline and Pfizer [16]. During the years 1995-2001, researchers at GSK identified more than 350 genes of interest from S. aureus, Streptococcus pneumoniae and H. influenzae by comparative genomic analysis. They found that out of 350 selected genes, only 127 genes are essential in at least one of these microorganisms. Of these, 67 were purified and tested by HTS against different libraries ranging from 260,000 to 530,000 compounds which yielded 16 hits, of which five resulted in a lead. Only one of these five leads (AFN-1252, compound 8, Fig. 3) further progressed to development as a fatty acid biosynthesis inhibitor. Fig. 2B schematically illustrates the development of AFN-1252 as a lead following the genomic approach. This compound inhibits the enoyl-acyl carrier protein (ACP) reductase (Fabl) and reduces the growth of all clinical isolates of S. aureus and S. epidermidis. However, AFN1252 has a narrow spectrum of antibacterial activity as the enoyl-ACP reductase is encoded by distinct enzyme isoforms in different bacterial species (e.g. Fabl in S. aureus and or FabK in S. pneumoniae [47] or FabV in P. aeruginosa [48]. The program was out licensed to Affinium. The candidate AFN1252 showed excellent tolerability and efficacy in a phase I clinical trial for oral treatment of S. aureus infections [49]. More recently, Affimium Pharmaceuticals completed the first oral Phase II clinical study in 2011 [50, 51]. The FDA in November 2013 designated AFN-1252 as a qualified infectious disease product (QIDP) affording beneficial regulatory incentives. In February 2014, AFN-1252 was acquired by Debiopharm™. Similarly, Pfizer reported four leads from their 65 HTS programs; however none of them progressed to clinical trials. In addition, Cubist pharmaceuticals focused on a family of enzymes called aminoacyl-tRNA synthetases. They have screened 17 enzymes of this family against 50,000 compounds but without success [52]. Despite of this positive evolution, the question remains why the genomic strategy was not as successful as anticipated? 3.2.1. Lessons learned from the genomic era regarding the targets 9 In the last few years, we have gathered considerable knowledge which explains why target-based HTS is not as successful as predicted. The reasons are mainly related to the selection of the targets themselves, the applied screening procedures and the libraries of compounds used as the source of inhibitors. The perception that the genomic revolution would reveal a treasure of novel, unexplored broadspectrum targets which would accelerate the process of antibiotic discovery, turned out to be wrong. Besides the criteria used for target selection like target conservation, evolutionary distance to human homologs and indispensible nature of the target, a target must fulfill several other and equally important requisites. The targets need to be validated in every species. For example, while screening quinolinone derivatives as MetRS inhibitors against 101 S. pneumoniae clinical isolates, researchers at GSK found that 20% of S. pneumoniae strains are intrinsically resistant due to the presence of a MetRS isoform [53]. Essentiality of the target must be validated under in vitro as well as in vivo conditions (in the nutrient rich environment of the human body) and it needs to be ascertained that the bacteria have no mechanism for circumventing the target reaction by acquiring its reaction product from the host. In recent studies it has been found that Gram-positive pathogens with low GC content were resistant to inhibitors of fatty acid biosynthesis type II in vitro, when medium is supplemented with exogenous fatty acids [54]. Examples of fatty acid inhibitors include AFN-1252 (GSK/Affinium) (compound 8, Fig. 3) [51], plantensimycin (compound 9, Fig. 3) [55], platencin (Merck) [56] and pyrroline diones (Bayer) [57]. In addition, deletion of essential fatty acid biosynthesis genes does not diminish the virulence of Streptococcus agalactiae in mice which further questioned the essentiality of the target under in vivo infection conditions [54]. Moreover, the new antibiotics should have a low tendency to provoke development of bacterial resistance due to a single point mutation. In summary, validation of the essential nature of the novel target for all species under the in vitro conditions as well as in the host (during the infection process) is mandatory. Although the techniques of in vivo target validation have been described, they are only used intermittently. Moreover, in the absence of a model target inhibitor, validation of the essentiality of the target becomes methodologically challenging which is often the case with novel or unexplored targets. 3.2.2. Lessons learned from the screening strategies The target selection and validation are only one part of the problem. The screening strategies themselves also play an important role in the success of this approach. When the compounds are tested against an isolated (cell-free) target, penetration through the bacterial cell wall is not an issue. As a result, the majority of the hits obtained turn out not to be able to penetrate the cell wall and thus will not exert antibacterial effect [16]. A way to overcome this problem is the design of whole-cell assays 10 that allow measurement of the target function in intact bacterial cells. For example, an assay involves differential sensitivity of target-depleted strains where the target can be silenced (by siRNA) or down regulated (by reduced promoter expression). In both cases, an inhibitor will exhibit differential antibacterial activity against mutant versus wild type strains. However, this technique leads to doubling of the screening efforts as it involves two detection points (i.e. for mutant and wild type strain) for each time point and inhibitor concentration. 3.2.3. Lessons learned from the compound pool Our inadequate knowledge about the bacterial physiology and suboptimal screening strategies does not explain the lack of success in almost all antibacterial high-throughput target-based screening campaigns. The problem also resides within the compound libraries. It is now widely accepted that the Lipinski’s ‘rule of five’ is irrelevant for antibiotics when coming from natural sources [58-60]. Moreover, the necessity to penetrate through the bacterial cell wall leads to physicochemical properties which are significantly different from that of other drugs [60]. In general, it has been observed that antibiotics are more hydrophilic and high molecular weight compounds. However, the dilemma is that most of the compound libraries from big pharma companies follow the Lipinski’s ‘rule of five’ (average physiochemical properties needed for all therapeutic area). As a result only a small subset of compounds is screened for antibacterial activity. Therefore, it can be concluded that the antibacterial screening approach should not be limited to compounds which follow Lipinski’s ‘rule of five’, rather it should consider the more natural product-like physicochemical properties [16]. 3.2.4. The valuable contribution of different “omics” strategies Overall, the genomic era should be recognized for its valuable contributions in our never ending fight against microbes. While indeed the genomic approach did not deliver what was originally anticipated in terms of new targets and/or new lead compounds, the contribution of genome mining, proteomics, comparative genomics and expression profiling and analogous techniques cannot be underestimated and has helped us gain a better understanding of antimicrobial action and of resistance mechanisms. Genomic differences between antibiotic-susceptible versus resistant strains can help to understand resistance mechanisms at the molecular level, which in turn helps to find additional targets to combat resistance. This could lead to the development of new antimicrobial compounds or co-drugs which enhance each other’s activities. This in turn will avoid or postpone the development of resistance [61, 62]. Genome profiling also taught us that antibiotics can have a differential effect on bacterial cells with in contrast low concentrations having a stimulating and differentiating effect. Herein, some antibiotics used at sub-inhibitory concentration even induce biofilm formation. This bacterial signaling mediated by antibiotics is not discussed here but has been nicely covered by Romero et al. [63]. 11 3.3. Targeting Virulence Factors: to Kill or Not to Kill? With the emergence of multidrug resistance, it is not sufficient to target only essential enzymes in the biosynthetic pathways, rather one should consider the virulence factors as a target for antiinfectives [64, 65]. The idea behind this strategy is not to kill but rather to hinder the pathogen so as to cause harm to the host. This is a ‘kinder and gentler’ approach in antibiotic discovery [65]. This approach is debatable because the virulence factors are important for causing harm to a host but not essential for the survival of pathogens [66]. Moreover, the progress in the field has been hampered by the difficulties in development of an in vitro assay to screen inhibitors. On the contrary, virulence factors are attractive targets for a number of reasons. Firstly, being outside the infected cell, targeting virulence factors would circumvent the problem of bacterial cell penetration. Secondly, these targets are absent in the host which is an added advantage. Thirdly, there is a possibility that compounds that target a ‘non-essential’ process are less prone to develop resistance [67]. Lastly, virulence specific therapeutics would have minimal impact on human gut flora. Although there are several potential advantages in targeting virulence factors, questions about the utility of such inhibitors as prophylactic agents, solo therapeutics or in combination with current antibiotics are always argued [65, 68, 69]. With the current improvements in the screening methodologies using genetically modified strains, there is hope for new and effective anti-virulence drugs [70]. In fact, the potential of anti-virulence agents as effective antibacterial agents is demonstrated by the positive results for the monoclonal antibodies against Clostridium difficile toxin A [71] and E. coli Shiga-like toxin 2 [72], although this overview does not intend to include passive immunotherapy. However, targeting virulence factors is more challenging due to our limited knowledge about the evolution of virulence factors and their roles in the bacterial physiology [73]. Here below, quorum sensing (QS) and the two components signal transduction (TCST) system have been described as a possible target. In general, virulence agents are responsible for the symptoms of an infection, allowing bacteria to adapt to the host environment. Therefore, one can also aim to modulate the production of these agents instead of directly inhibiting their action. Hence, blockade of virulence gene expression can be envisaged and has been shown to significantly attenuate infections by disarming bacteria. This gene regulation strategy for virulence factors has been adequately documented by Williams and colleagues and will not be discussed further [74, 75]. 3.3.1. Quorum sensing: an unexplored therapeutic strategy Before discussing about QS inhibitors as potential antibacterial agents, a brief overview of QS is necessary. QS is a fundamental process of bacterial cell-cell communication by producing and responding to small diffusible molecules that act as signals. These small molecules are called 12 autoinducers (AIs) [76, 77]. AIs are produced in response to the changing population density to coordinate the expression of virulence factors in both Gram-positive and Gram-negative bacteria. There are three types of AIs that have been identified so far: N-acylated homoserine lactone (acyl-HSL), autoinducing peptide (AIP) and the boronate ester autoinducer-2 compounds (AI-2). In Gram-negative bacteria, acyl-HSL acts as an autoinducer whereas small peptides (AIP) play the same role in Grampositive bacteria [78, 79]. AI-2 is an interspecies autoinducer. Representative structures for all three types of AIs are depicted in Fig. 4 (compound 10-12). The LuxI/LuxR bioluminescence system of Vibrio fischeri is the most intensely studied QS system and will be discussed here as representative example. As shown in Fig. 5 [80], bacteria produce diffusible AIs (e.g. acyl-HSL) via the inducer synthase (LuxI-type protein). As cell population density increases, the concentration of AI inside and outside the cell increases. At critical AI concentration, an autoinducer receptor protein (LuxR) binds to the AI. The AI-LuxR complex most often homodimerizes and binds to adjacent QS promoter (luxICDABE, termed as ‘lux boxes’), which then activates a variety of group behavior such as expression of virulence factors, swarming and biofilm formation, besides many other activities [67]. As QS plays a vital role in establishing infections by most of the pathogens, interception of QS represents an ideal strategy to disarm the pathogen. The QS system can be targeted at three points: suppressing the acyl-HSL production (LuxI), inactivating the signal molecule (AI) and blocking the receptor (LuxR). Inhibition at any one of these points is expected to lead to a bacterial communication breakdown. 3.3.2. QS inhibitors 3.3.2.1. Targeting the synthase (I) Acyl-HSL synthases are considered as ideal antimicrobial targets as they are absent in eukaryotes and they are essential for QS. Inhibition of synthase protein appears a straightforward approach, as cellcell communication is impossible without AI. However, as these crystal structures of LuxI from different species were reported more than a decade ago [81, 82] there are only very few reported studies that specifically target LuxI [83]. Moreover, measuring the enzymatic activity is also cumbersome. Lately, a few synthase inhibitors have been identified from HTS [84]. In Burkholderia glumae wild type (wt) N-octanoyl-homoserine lactone (C8-HSL) is synthesized by TofI (acyl-HSL synthase from B. glumae). In 2011, Rhee et al. screened a library of 55 compounds for inhibition of QS-mediated production of toxoflavin and C8-HSL and identified two hits along with a few minor hits. The natural AI (C8-HSL, 13) and the hits J8-C8 (14) and E9C-3oxoC6 (15) are depicted in Fig. 6. Although both compounds inhibited the synthesis of C8-HSL and also inhibit QS-dependent ahpF expression, E9C3oxoC6 was shown to be more potent than J8-C8. Detailed mechanistic studies suggest that the 13 compound J8-C8 inhibits TofI (C8-HSL synthesis) whereas E9C-3oxoC6 competitively inhibits binding of C8-HSL to the toxoflavin receptor (TofR). 3.3.2.2. The acyl-HSL inactivation Degradation or inactivation of the signal molecule would be a complementary strategy to combat the QS dependent virulence factor expression. Indeed, several organisms including prokaryotes and eukaryotes are known to degrade the acyl-HSL signal in order to block QS of invading pathogens. There are several ways by which degradation of acyl-HSL can take place such as increase of pH at the infection site (in certain plants species) provoking hydrolysis of the lactone, secretion of oxidized halogenated compounds which can react with acyl-HSL leading to inactive derivatives, secretion of degrading enzymes such as lactonase or acylase, use of acyl-HSL as carbon and nitrogen source. Although acylHSL inactivation according to current views does not represent a viable strategy in human and animals, it can have significant value for agricultural application. For example, transgenic plants producing acylHSL lactonase have shown to be resistant to infections caused by several Gram-negative plant pathogens [85]. 3.3.2.3. Targeting the receptor (R) The third target in the QS system is the receptor (LuxR type) protein. As compared to the synthase, it is a widely studied medicinal chemistry target with significant genetic, biochemical and structural data being available. In 1986, Eberhard et al. [86] evaluated ~20 acyl-HSL analogues for their agonistic and antagonistic activity against V. fischeri B-61 (a strain that produces more light with addition of exogenous 3-oxo-acyl-SHL). The authors concluded that the -lactone ring is a prerequisite for LuxR agonistic activity whereas replacement of the -lactone ring with the thiolactone or lactam yielded compounds with inhibitory or no activity. Secondly, the optimal chain length for agonistic activity was six carbons and deviating from this length resulted in inhibitory activity, with a nine carbon long side chain being the optimum for antagonistic activity [87]. Moreover, 3-oxo groups were claimed to be important for potency. The most active compounds of the series are shown in Fig. 7 (structures 16-18). A decade later, in 1996, Greenberg et al. [87] evaluated the activity of acyl-HSL analogues in the E. coli reporter strain VJS533 pHV200I, which harbors LuxR and V. fischeri luminescence gene cluster with luxI inactivated. This study revealed the same results as reported by Eberhard et al. [86]. More recently, De Keersmaecker et al. [88] reported different strong activators of a LuxR homologue from Salmonella enterica and of V. fischeri. In 2002, inspired by halogenated furanone modulators of QS, Reverchon et al. [89] reported a series of acyl-HSL analogues having various substitutions on the 3-and 4-position of -lactone ring and 14 studied their activities against LuxR. The authors discovered that substitution on the 4-position resulted in decreased agonistic activity whereas substitution at the 3-position is well tolerated. In fact, the compounds 19 and 20 (Fig 7) act as antagonists at high concentration. Moreover, R stereochemistry at the 3-position (compound 21, Fig. 7) is important for agonistic activity while inversion of stereochemistry resulted in a 1000-fold reduced activity. The authors concluded that branched alkyl or cycloalkyl acyl-HLSs are potent agonists, with notably cyclohexyl acyl-HSL being as potent as the natural ligand [89]. Further, replacement of the acyl-HSL amide with sulfonyl or urea moieties resulted in compounds that were potent LuxR inhibitors [90, 91] (see Fig. 7 compounds 22 and 23). In 2008, Blackwell et al. [78] revealed several modulators of LuxR ranging from agonist to antagonist. They found that phenylacetanoyl homoserine lactones (PHLs) with an electron withdrawing group at the para-position of the phenyl ring were the most potent inhibitors of LuxR (compound 24, Fig. 7). Lately, Wang et al. [92-94] reported a series of AI-2 quorum sensing inhibitors through structure-based virtual screening. Representative examples of these sulfone and adenosine derivatives are depicted in Fig. 8 (compound 25-30). A recent and extensive review on acyl-homoserine lactone quorum sensing by Greenberg et al. [95] is worthwhile consulting for this specific topic. 3.3.3. Challenges in the QS targeting strategy There are many hurdles while considering QS as a therapeutic target. Development and standardization of the assay method is one of the largest hurdles in QS targeting. It has been found that not all compounds display similar activity versus different strains. This lack of compatibility can be overcome by using an in vitro assay with purified protein. However, the progress has been hampered due to difficult purification and manipulation of these proteins in the absence of a natural ligand. Researchers have used invertebrate or plant species as a surrogate model to study bacterial pathogenesis [96-98]. While these surrogate models cannot be used to address pharmacokinetic issues, they can be used for rapid screening of large compound libraries [99, 100]. Moreover, it has been shown that QS mutant strains of P. aeruginosa displayed reduced virulence as compared to wild-type P. aeruginosa, but none of the mutations resulted in avirulent strains [67, 85, 101]. This suggests that besides QS, other factors also play a key role in infection [102]. Finally, QS is a new and rapidly expanding field and we have limited knowledge about the mode of action of the QS inhibitors. 3.3.4. Two-component signal transduction systems (TCSTS) The bacterial two-component signal transduction system is responsible for sensing the extracellular environment and for responding accordingly so that the bacteria can survive. The essence 15 of TCSTS lies in the recognition and interpretation of external signals and conversion of those signals into transcription activation or repression of specific genes. It is one of the important systems in which information is processed. The factors regulated by TCSTS include host invasion, drug resistance, motility, phosphate uptake, osmoregulation, nitrogen fixation and other functions. Bacterial TCSTS typically comprise of a membrane-bound histidine kinase (HK, sensor kinase) and a cytoplasmic regulator [103]. In response to external signals, the HK catalyzes ATP-dependent autophosporylation on a histidine residue. The phosphoryl group is subsequently transferred to an aspartate residue on a cognate response regulator. Phosphorylation of the response regulator activates the DNA-binding properties of the C-terminal domain, resulting in expression modulation of the virulence factors (Fig. 9) [104]. A number of features of TCSTS make them a potential target for antimicrobial therapy. At First, significant homology exists among the HK and response regulator proteins, in particular around the active sites, so that TCSTS inhibitors can yield broad-spectrum antibiotics [105, 106]. Secondly, TCSTS is responsible for expression of virulence factors which are essential for survival of pathogens inside the host [107]. Furthermore the Ser/Thr kinase signaling pathways in eukaryotes are different from HK-mediated signal transduction in prokaryotes. This difference can be used for selective targeting of bacterial TCSTS [104]. 3.3.5. Inhibitors of TCSTS Understanding how TCSTS works, provides several potential points of intervention like signal sensing, autophosphorylation of the sensor kinase, phosphotransfer to the regulator, or dephosphorylation of the regulator. Biochemical assays used to screen potential inhibitors are based on the autophosphorylation reaction of HK and the subsequent phosphoryl transfer to the response regulator with ATP as the phosphate donor [108]. In recent years, some TCSTS inhibitors have been discovered by pharmaceutical industry through screening of large chemical libraries and a structure activity relationship (SAR) program of the resulting lead compounds. One representative structure of each chemical class found, is depicted in Fig. 10 (compounds 31-39). Among them, the compound RWJ-49815 (compound 31) and its derivatives are claimed to exhibit bactericidal activity against several Gram-positive bacteria. Their activity spectrum includes MRSA, VRE and penicillin resistant S. pneumoniae [104]. Most of the hits obtained via screening of large compound libraries often suffer from poor selectivity and specificity. However, with recent advancements in the structure determination of several proteins of TCSTS, the stage is now set for rational structure-based drug design. Finally, in a recent review Gordon et al. [74] discussed the medicinal chemistry perspective of attenuating virulence gene regulation for the possible treatment of staphylococcal infections. 16 3.4. Targeting Non-Multiplying Bacteria In clinical infections, bacteria exist in two different states which are described as multiplying (logarithmic phase) and non-multiplying (stationary, dormant or latent phase) [20, 109]. Most of the multiplying bacteria are killed by antibiotic therapy whereas non-multiplying or slowly multiplying bacteria can survive with repeated doses of antibiotics. Although, non-multiplying bacteria do not cause disease, they act as a pool for multiplying bacteria and are responsible for recurrence of infection. These non-multiplying bacteria are difficult to eradicate by antibiotics resulting in prolonged therapy, emergence of resistance and reduced patient compliance (e.g. tuberculosis (TB) and leprosy) (Fig. 11) [20]. A possible solution to this problem, which at present is mostly related to mycobacterial infections, could be to target multiplying and non-multiplying bacteria simultaneously. This strategy in theory should shorten the duration of therapy, slow down or avoid emergence of resistance, decrease the cost of therapy and increase the patient compliance. 3.4.1. Existing antibiotics and new developments against non-multiplying bacteria There are few marketed antibiotics known to kill non-multiplying bacteria more effectively than others and these are found among the inhibitors of mycobacterial infections. Examples of these antibiotics include pyrazinamide (PZA, 40, Fig. 12), moxifloxacin (41, Fig. 12) and rifampicin (42, Fig. 12). The antitubercular activity of pyrazinamide is pH dependent. Recent studies have shown that pyrazinamide enters Mycobacterium tuberculosis by passive diffusion. It is then metabolized by nicotinamidase or pyrazinamidase to pyrazinoic acid (POA) which is then excreted by a weak efflux pump. Under acidic conditions, protonated POA (HPOA) is reabsorbed and accumulates in the cell (due to the inefficient efflux pump) leading to increased acidity and cell death. Despite of excellent in vivo activity, PZA is inactive against M. tuberculosis under in vitro or culture conditions close to neutral pH. Furthermore, it has no bactericidal activity against rapidly growing bacilli in patients during the first two days of treatment [110, 111]. Addition of moxifloxacin and gatifloxacin to the regimen of rifampicin, pyrazinamide and isoniazid increases the death of a non-multiplying culture of M. tuberculosis [112]. These results motivate researchers to search for new antibiotics targeted to nonmultiplying bacteria and aimed at shortening treatment, increased patient compliance and cure rates. In the last eight years, five drug candidates have entered clinical trials for the treatment of genetically resistant strains of M. tuberculosis. Chemical structures of these drug candidates are given in Fig. 13. These drug candidates include PA-824 (compound 43, Fig. 13) (Pathogenesis/Chiron and GATB), [113] diarylquinolone R207910/TMC 207 (compound 44, Fig. 13) (Tibotec/Johnson and Johnson) [114], nitroimidazole OPC-67683 (compound 45, Fig. 13) (Otsuka Pharmaceuticals) [115], diamine SQ109 (compound 46, Fig. 13) (Sequella Inc and NIH) [116] and Pyrole LL 3858 (compound 47, Fig. 13) (Lupin Ltd) [117]. 17 PA-824 (compound 43, Fig. 13) is a nitroimidazole derivative with a complex mode of action which is active against both multiplying and non-multiplying bacteria (M. tuberculosis). Under aerobic condition, it acts by inhibiting the cell wall biosynthesis (i.e., the mycolic acid part) through an unclear mechanism whereas under anaerobic condition, it acts as an oxide nitric (NO) donor that causes respiratory poisoning through formation of nitric oxide. Recently it has been tested in a Phase II clinical trial for treatment of resistant TB and was shown to be safe, well tolerated and effective at doses of 100200 mg/day [118]. Bedaquiline (TMC 207, compound 44, Fig. 13) belongs to the diarylamine class and has a unique mode of action inhibiting the ATP-synthase. It is currently in a phase III trial for treatment of patients with pulmonary multidrug resistance (MDR)-TB. The compound OPC 67683 (compound 45, Fig. 13) likewise is under phase III clinical evaluation for treatment of MDR-TB. It acts by inhibiting the synthesis of methoxy and keto-mycolic acid biosynthesis. SQ 109 (compound 46, Fig. 13) was identified through random screening of large chemical libraries based on a 1,2-diethylamine scaffold. Phase I and Phase II data suggest it to be safe and well tolerated. Although the pyrrole LL 3858 (compound 47, Fig. 13) showed good efficacy in a mouse model (12.5 mg/Kg, for 12 weeks), its mode of action is unclear. Extensive research likewise has been ongoing for the membrane-active agents XF-70 (compound 48, Fig. 13) and XF-73 (compound 49 Fig. 13) [119], which showed nice bactericidal activity against slow-growing and non-dividing cultures of S. aureus, including against biofilms. With HT61, a small quinolone-derived compound (undisclosed structure) has been heralded recently being strongly active against non-multiplying bacteria as well as against mupirocin resistant MRSA [120]. It effectively kills these non-multiplying cells very rapidly by permeabilization of the cell membrane and destruction of the cell wall. Synergistic activities were noticed upon combination with either neomycin, gentamicin or chlorhexidine [121, 122]. 3.4.2. New molecular targets in non-multiplying bacteria In spite of the complex signaling pathway regarding virulence gene expression, and the multitude of effects that these virulence factors can have, some research groups have tried to identify specific genes responsible for antibiotic tolerance in non-multiplying bacteria. Uncovering such a gene could make it possible to develop an inhibitor interfering with its gene product thus rendering non-multiplying bacteria susceptible to antibiotics. E. coli has been studied extensively in search for such target in nonmultiplier bacteria. These studies revealed that bacteria express the sigma factor RpoS (RNA polymerase sigma S, main regulator of stationary phase genes) when reaching the stationary phase. This factor is responsible for decreased drug sensitivity in an acrAB mutant (inactive efflux pump) [123] which implies that RpoS might be involved in multidrug resistance. Deletion of rpoS in P. aeruginosa results in a mutant which is much more sensitive to antibiotics than the wild-type, confirming this hypothesis [124]. The phosphate regulon (PhoU) is also an interesting target and believed to be involved 18 in tolerance to antibiotics and stress. Deletion of phoU in E. coli results in increased susceptibility of the mutant as compared to wild-type, in both multiplying and non-multiplying bacteria. PhoU is proposed to regulate cell mobility, nutrient transport and phosphate and energy metabolism, functions which are all linked to antibiotic tolerance. Another potential target is the penicillin-binding protein 1b (PBP1b) [125], or the intergenic region between aldB (aldehyde dehydrogenase B) and yiaW (inner membrane protein) [126], which both also are linked to antibiotic tolerance [127]. 3.4.3 Challenges in targeting non-multiplying bacteria Although, theoretically, targeting non-multiplying bacteria seems a promising and attractive strategy, there are several important issues which need to be addressed. Above mentioned antibiotics are relatively ineffective at killing non-multiplying bacteria. In fact, none of these drugs (either marketed or in clinical trials) have been screened using whole non-multiplying bacteria or molecular targets that are essential for bacteria in their dormant state. This approach is less viable due to poor penetration of compounds into the bacterial cell and there might be a limited number of targets due to low or no metabolic activity [123, 128]. Secondly, each microorganism has many non-multiplying forms. For example, persisters contribute only 1% of the non-multiplying population, and were found to have a different expression profile than log phase and non-multiplying bacteria (with “persisters” referring to genetically drug susceptible quiescent (non-growing or slow growing) organisms that survive exposure to a given antibiotic or drug and have the capacity to revive (regrow or resuscitate and grow) under highly specific conditions [129]). Here the question which needs to be addressed is which form of non-multiplying bacteria should be targeted [130]? It is still not clear which forms of in vitro non-multipliers are equivalent to those found in the host during clinical infection [131]. Finally, standard tests need to be developed for comparing antibiotic potency [132]. More recently, however, a new milestone was reached by the research group of Den et al. [133, 134], who succeeded in reducing the persistence during the growth of both E. coli and P. aeruginosa and to restore the antibiotic susceptibility of persister cells using the quorum sensing inhibitor (Z)-4bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one (BF8, compound 50, Fig. 13). Persister control was noted for both planktonic and biofilm cells for both bacteria. While this compound showed potent activity in persister control, it could not be determined whether this was through genuine QS inhibition or whether the effect is mediated by another target. The latter seems plausible as a similar effect of reverted antibiotic tolerance was noted for an E. coli luxS mutant. 3.5. Bacteriophage Therapy: Old Dogma New Tricks? In the early part of 20th century, Frederick Twort and Felix d’Herlle discovered bacteriophage independently [135]. Bacteriophages or simply phages are viruses with an ability to infect and kill bacteria, which forms the basis for using phages as therapeutic agents [136, 137]. Immediately 19 following their discovery, bacteriophages have been used clinically during the pre-antibiotic era (19201930s) [138]. The use of bacteriophages as biological antibacterials has persisted globally. This includes former Soviet Republic of Georgia, Poland, France, Russia, United States, etc. Use of phages as therapeutic agent offers several advantages. Firstly, phages are bacterium specific, attacking specific bacteria without affecting normal microflora. Secondly, phages replicate naturally and exponentially and are therefore effective in treating infections. Finally, they are easy to prepare and to use and show no adverse effect when used in a cocktail (multi-component phage preparation). 3.5.1. Limitation of phage therapy The use of bacteriophages as antibacterial agents was abandoned due to discovery of broadspectrum antibiotics coupled with other factors like poor understanding of phage biology, shortage of data from clinical trials and lack of documentation at that time. Despite of their advantages, there are several key issues regarding the use of phages as antibacterial agents. Firstly, phages are pathogen specific, therefore displaying a narrow spectrum of activity which limits their use as therapeutics in absence of a confirmed diagnosis of a specific pathogen. Secondly, in order to use a phage preparation effectively, it is necessary to have a set of well characterized phages available for a broad-range of bacterial pathogens [139]. Thirdly, there are serious difficulties in standardizing protocols for preparation and quality control. Further, when used systemically, phages may induce an immunogenic response or neutralizing antibodies. Finally, there is the potential of triggering a toxic shock due to massive bacterial lysis [140, 141]. Despite of these disadvantages, phage therapy was part of the standard healthcare system in the Soviet Union even during the 1960s and 1970s. In former Soviet Union, phage preparations were used for therapy, prophylaxis and diagnosis of many bacterial infections [142]. In 1990, Abdul Hassan et al.[143] documented the use of phage therapy for the treatment of 30 cases of burn wound associated antibiotic-resistant P. aeruginosa sepsis. In 18 of 30 cases there was good (12 cases, 60-70% recovery) or even excellent (6 cases with >90% recovery) improvement in skin graft take [143]. However, the authors recommended phage therapy should be restricted to antibiotic-resistant strains. In 2005, Jikia et al. [144] reported the use of bacteriophage to treat 90Sr radiation burns suffered by three lumberjacks from Georgia. Subsequently, the burns became infected with MRSA. After unsuccessful treatment with antibiotics for one month, phage therapy was attempted using PhageBioDerm along with ciprofloxacin. Following 2 days of treatment, purulent drainage decreased to almost none and testing failed to detect MRSA on the seventh day. As no negative controls were employed, this study does not provide proof-of-concept for phage therapy. Similarly positive outputs of phage therapy have been reported by Lazareva et al. [145]. In 2007, Fralick et al. [146] studied the effect of a phage cocktail in treatment of P. aeruginosa infection in a mouse burn wound model. In this 20 study, a number of 2x102 to 3x103 rifampicin-resistant P. aeruginosa (POA1Rif) were injected at the burn site. A phage cocktail inoculum (~3x108/100 μL) was administered by intraperitoneal (i.p.), intramuscular (i.m.) or subcutaneous (s.c.) route, respectively. The authors observed that not all treated mice did survive and that the route of phage administration played an important role in the treatment efficacy with the i.p. route providing maximum protection (87%) [146]. Herein, “lysis from without” or a non-productive phage infection kills bacteria as of its high multiplicity of virion adsorption [138]. This is an important mechanism of killing bacteria via high dosing regimens, but is not the strategy which should be aimed for in view of a possible immunogenic response or toxic shock. Moreover, in view of concerns for an immune response even with a lower inoculum followed by the normal “lysis from within” induced by phage proteins, Paul et al. [147] in contrast developed lysis-deficient phages by disruption of the endolysin gene. These still kill the target cells but are incapable of host cell lysis, but such recombinant phage still proved lethal to methicillinresistant S. aureus [147]. A recent review paper also discusses the biocontrol of bacterial infections using phage therapy from a physiological pharmacology standpoint [148]. Moreover, even today, scientists of the military hospital in Brussels and of KU Leuven are joining force to evaluate and use phages for the treatment of severe burns [149]. Recently, use of phage therapy to treat burn wound was evaluated in a small phase I study at the Burn Wound Centre of the Queen Astrid Military Hospital, Brussels, Belgium. A small group of 9 patients were treated with the BFC-1 phage cocktail containing a mixture of 3 lytic phages: Myovirus, Podovirus against P. aeruginosa and Myovirus against S. aureus. In the study, a large burn section was exposed to a single spray application of a phage cocktail while a distant portion of the wound was taken as control. The complete results of this study are yet to be published. However, a clear legal framework for bacteriophage therapy is largely lacking and this forms the bottleneck to establish this methodology as a valid treatment option [150, 151]. 4. CONCLUSIONS With the emergence of multidrug resistance, there is an urgent need for new antibacterial agents. Chemical modifications of existing antibiotics yielded short term success with rapid development of cross resistance among the same class of antibiotics. Moreover, the success in discovering antibiotics through a conventional approach is limited (only two new classes of antibiotics entered the market in last 50 years). The SBDD approach has been successfully used for the design of a DHFR inhibitor (iclaprim) where the molecular basis of resistance has been utilized for antibiotic design. Phase III clinical trials demonstrate that iclaprim is well tolerated and effective in treating complicated soft structure skin infection which clearly shows that SBDD is a powerful tool for antibiotic discovery. Moreover, SBDD also successfully has been used for the discovery and optimization of 21 ligands for the targets such DNA gyrase subunit B, LpxC, MetRS, etc. Targeting of Lipid II by the HNP1 defensin small molecule analogues recently uncovered (BAS00127538), seems a valuable strategy for development of broad-spectrum therapeutics as glycopeptides like vancomycin already established the essentiality of Lipid II. Likewise, the heterocyclic sulfonamide analogues as inhibitors of ThrRS and studied by Trius Therapeutics hold potential in view of their high activity and bacterial selectivity. While they permeate cellular membranes, the antibacterial activity remains mediocre. Moreover, they also prove efficient substrates for efflux pumps and further optimization of these compounds is needed to improve their potency. Although the genomic revolution provided us with a treasure of targets known to be essential for the bacterial survival, it is not clear yet how many of them are druggable targets. The genomic revolution is just beginning and it therefore is too early to comment on the success of the genomic approach. Moreover, validation of the novel target, screening strategies and compound pools also contribute to failures with the genomic approach in terms of establishing new targets or new leads. However, comparative expression profiling has thoroughly increased our understanding of resistance mechanisms and of biofilm formation, which will assist us in developing improved antibiotics. Anti-virulence strategy is often debatable due to the non-essential nature of the virulence factors. Targeting virulence factors, either directly or via modulation of their gene expression, seems a promising strategy as demonstrated by positive results of clinical trials for monoclonal antibodies against C. difficile toxin A and E. coli Shiga like toxin 2. Moreover, with current improvements in the screening methodologies, there is hope for a new and effective anti-virulence drug. However, further research efforts are needed to get insight into the mechanism by which the expression of a virulencerelated gene is activated or inhibited and the development of small molecule inhibitors for modulation of virulence gene expression has a long way to go. The same holds for the different strategies to inhibit quorum sensing: some nice activities have been obtained in the test tube, but many challenges remain before quorum sensing will be established as a viable target for clinically related infections. Theoretically, targeting multiplying and non-multiplying bacteria simultaneously can shorten the duration of therapy, decrease the chances of development of resistance and also reduce the cost for therapy and increase the patient compliance. However, further clinical relevance of an in vitro inhibitor of a non-multiplier is still lacking. Recently, five drug candidates have shown effectiveness for treating MDR-TB and are at various stages of clinical trials. While persistence is vital problem in antibacterial therapy, recent efforts succeeded in restoring the sensitivity to antibiotics using a quorum sensing inhibitor. If the latter finding can be confirmed this might prove to be a new milestone in the continuous fight against bacteria. Bacteriophages, a revisited treatment modality, offer several advantages which make them attractive therapeutic agents. The positive outcome of many previous studies suggests that phage 22 therapy, if properly applied, can play a vital role in controlling infectious diseases which remain otherwise untreated. Herein, genetically modified bacteriophages like the lysis-deficient phages recently developed maybe could remediate for the missing legal framework for therapeutic use of phages. Although none of the discussed approaches (either old or new) has potential to discover new resistance-breaking antibiotics solely, we must consider applying two or more approaches to improve the antibiotic discovery process at every stage (e.g. the genomic approach can be used for validation of the target, HTS for rapid screening of large chemical libraries, SBDD for optimization of a lead, etc). Besides the discovery of new antibiotics, efforts should also be directed to increase the hygiene, to reduce misuse or abuse of antibiotics and self-medication. Moreover, a combination of drug regimens likewise is very effective to reduce the chances of resistance development. Finally, we wish to come back briefly on our economic statements put forward in the introduction. As the financial reward for developing new antibacterials was inadequate, American and European government agencies have wisely implemented additional incentives among which improved IP protection to drive innovation. Whether this will be sufficient remains to be seen, but the overall cost for developing new therapeutic classes is skyrocketing. Most researchers aim for and hope to find broad spectrum antibiotics also for economic reasons, allowing more patients to be treated. Wider use at the same time however increases the chances for resistance development, especially if only a single cellular target is under attack. Combination therapies like with HIV and HCV antivirals maybe could become standard when treating serious infections, but at which economic cost? Narrow spectrum antibiotics therefore should be reserved for treating deadly infections in an intensive care unit as a last resort therapy and to avoid resistance generation. Obviously, sufficient remuneration to compensate for the limited therapeutic use should be implemented. However, society can no longer carry the exuberant costs as charged for some antivirals, with a multitude of highly specialized treatments coming up for fighting different cancers, viruses, bacteria and rare diseases. Finding the right balance between adequate incentives and remuneration versus the overall cost for drug development will need a lot of consideration in the near future. CONFLICT OF INTEREST Both authors declare no conflicts of interest are involved. ACKNOWLEDGEMENTS Bharat Gadakh thanks Erasmus Mundus External Cooperation Window lot 13 (EMECW13) and KU Leuven (GOA/10/13) for financial support. Antibiotic research is made possible by a grant 23 from the FWO (G.0778.14N; Flemish Government) and from the Research Fund KU Leuven (OT/14/105). We also thank all four reviewers and the guest editor for their constructive criticisms which considerably improved this review. ABREVIATIONS AIP = Autoinducing peptide AIs = Autoinducers ATP = Adenosine triphosphate cSSSI = Complicated soft structure skin infection DHFR = Dihydrofolate reductase HK = Histidine Kinase HSL = Homoserine lactone HTS = High throughput screening MDR-TB = Multidrug resistant tuberculosis MIC = Minimum inhibitory concentration MRSA = Methicillin resistant Staphylococcus aureus NCEs = New chemical entities NMR = Nuclear Magnetic Resonance PHL = Phenylacetanoyl homoserine lactone POA = Pyrazinoic acid PZA = Pyrazinamide QIDP = Qualified infectious disease product QS = Quorum sensing SAR = Structure activity relationship SBDD = Structure-based drug design siRNA = Small interfering RNA SMX = Sulfamethoxazole TB = Tuberculosis 24 TCSTS = Two components signal transduction system TMP = Trimethoprim VRE = Vancomycin Resistant Enterococcus REFERENCES [1] Coates, A.R.; Hu, Y. Novel approaches to developing new antibiotics for bacterial infections. Br. J. Pharmacol., 2007, 152, 1147-1154. [2] Brotz-Oesterhelt, H.; Sass, P. Postgenomic strategies in antibacterial drug discovery. Future Microbiol., 2010, 5, 1553-1579. [3] Fakuda, Y. New approaches to overcoming bacterial resistance. Drugs Fut., 2009, 34, 127136. [4] Bockstael, K.; Van Aerschot, A. Antimicrobial resistance in bacteria. Cent. Eur. J. Med., 2009, 4, 141-155. [5] Charifson, P.S.; Grossman, T.H.; Mueller, P. The use of structure-guided design to discover new anti-microbial agents: focus on antibacterial resistance. Anti-Infect. Agents Med. Chem., 2009, 8, 73-86. [6] Projan, S.J.; Bradford, P.A. Late stage antibacterial drugs in the clinical pipeline. Curr. Opin. Microbiol., 2007, 10, 441-446. [7] Katz, M.L.; Mueller, L.V.; Polyakov, M.; Weinstock, S.F. Where have all the antibiotic patents gone? Nat. Biotech., 2006, 24, 1529-1531. [8] Talbot, G.H.; Bradley, J.; Edwards, J.E.; Gilbert, D.; Scheld, M.; Bartlett, J.G. Bad bugs need drugs: An update on the development pipeline from the antimicrobial availability task force of the infectious diseases society of America. Clin. Infect. Dis., 2006, 42, 657-668. [9] Simmons, K.J.; Chopra, I.; Fishwick, C.W.G. Structure-based discovery of antibacterial drugs. Nat. Rev. Microbiol., 2010, 8, 501-510. [10] Overbye, K.M.; Barrett, J.F. Antibiotics: Where did we go wrong? Drug Discov. Today, 2005, 10, 45-52. [11] Senior, M. Sovaldi makes blockbuster history, ignites drug pricing unrest. Nat. Biotech., 2014, 32, (6), 501-502. [12] Kroll, D., Antibiotic pipeline revival: FDA approves Cubist Pharmaceuticals' sivextro for MRSA, other serious skin infections. Forbes 20/06/2014, 2014. [13] Edson, J.A.; Kwon, Y.J. RNAi for silencing drug resistance in microbes toward development of nanoantibiotics. J. Control. Release, 2014, 189, 150-157. [14] Brogden, K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol., 2005, 3, 238-250. [15] Pushpanathan, M.; Gunasekaran, P.; Rajendhran, J. Antimicrobial peptides: Versatile biological properties. Int. J. Peptides, 2013, 2013, 15. [16] Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov., 2007, 6, 29-40. [17] Gwynn, M.N.; Portnoy, A.; Rittenhouse, S.F.; Payne, D.J. Challenges of antibacterial discovery revisited. Ann. N. Y. Acad. Sci., 2010, 1213, 5-19. [18] Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov., 2013, 12, 371-387. [19] Isaacson, R.E. Novel targets for antibiotics. Expert Opin. Invest. Drugs, 1994, 3, 83-91. [20] Coates, A.; Hu, Y.; Bax, R.; Page, C. The future challenges facing the development of new antimicrobial drugs. Nat. Rev. Drug Discov., 2002, 1, 895-910. [21] Barker, J.J. Antibacterial drug discovery and structure-based design. Drug Discov. Today, 2006, 11, 391-404. [22] Anderson, A.C. The process of structure-based drug design. Chem. Biol., 2003, 10, 787-797. 25 [23] Congreve, M.; Murray, C.W.; Blundell, T.L. Keynote review: structural biology and drug discovery. Drug Discov. Today, 2005, 10, 895-907. [24] Kompis, I.M.; Islam, K.; Then, R.L. DNA and RNA synthesis: antifolates. Chem. Rev., 2005, 105, 593-620. [25] Canton, R.; Loza, E.; Morosini, M.I.; Baquero, F. Antimicrobial resistance amongst isolates of Streptococcus pyogenes and Staphylococcus aureus in the PROTEKT antimicrobial surveillance programme during 1999-2000. J. Antimicrob. Chemother., 2002, 50, 9-24. [26] Vickers, A.A.; Potter, N.J.; Fishwick, C.W.G.; Chopra, I.; O' Neill, A.J. Analysis of mutational resistance to trimethoprim in Staphylococcus aureus by genetic and structural modelling techniques. J. Antimicrob. Chemother., 2009, 63, 1112-1117. [27] Summerfield, R.L.; Daigle, D.M.; Mayer, S.; Mallik, D.; Hughes, D.W.; Jackson, S.G.; Sulek, M.; Organ, M.G.; Brown, E.D.; Junop, M.S. A 2.13 à structure of E. coli dihydrofolate reductase bound to a novel competitive inhibitor reveals a new binding surface involving the m20 loop region. J. Med. Chem., 2006, 49, 6977-6986. [28] Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol., 2006, 71, 941-948. [29] Krievins, D.; Brandt, R.; Hawser, S.; Hadvary, P.; Islam, K. Multicenter, randomized study of the efficacy and safety of intravenous iclaprim in complicated skin and skin structure infections. Antimicrob. Agents Chemother., 2009, 53, 2834-2840. [30] Breukink, E.; de Kruijff, B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov., 2006, 5, 321-323. [31] Leeuw, E.d.; Li, C.; Zeng, P.; Li, C.; Buin, M.D.-d.; Lu, W.-Y.; Breukink, E.; Lu, W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett., 584, 1543-1548. [32] Varney, K.M.; Bonvin, A.M.J.J.; Pazgier, M.; Malin, J.; Yu, W.; Ateh, E.; Oashi, T.; Lu, W.; Huang, J.; Diepeveen-de Buin, M.; Bryant, J.; Breukink, E.; MacKerell, A.D., Jr.; de Leeuw, E.P.H. Turning defense into offense: Defensin mimetics as novel antibiotics targeting Lipid II. PLoS Pathog., 2013, 9, e1003732. [33] de Leeuw, E.P. Efficacy of the small molecule inhibitor of Lipid II BAS00127538 against Acinetobacter baumannii. DDDT, 2014, 8, 1061. [34] Teng, M.; Hilgers, M.T.; Cunningham, M.L.; Borchardt, A.; Locke, J.B.; Abraham, S.; Haley, G.; Kwan, B.P.; Hall, C.; Hough, G.W.; Shaw, K.J.; Finn, J. Identification of bacteria-selective threonyl-tRNA synthetase substrate inhibitors by structure-based design. J. Med. Chem., 2013, 56, 1748-1760. [35] Tari, L.W.; Li, X.; Trzoss, M.; Bensen, D.C.; Chen, Z.; Lam, T.; Zhang, J.; Lee, S.J.; Hough, G.; Phillipson, D.; Akers-Rodriguez, S.; Cunningham, M.L.; Kwan, B.P.; Nelson, K.J.; Castellano, A.; Locke, J.B.; Brown-Driver, V.; Murphy, T.M.; Ong, V.S.; Pillar, C.M.; Shinabarger, D.L.; Nix, J.; Lightstone, F.C.; Wong, S.E.; Nguyen, T.B.; Shaw, K.J.; Finn, J. Tricyclic GyrB/pare (TriBE) inhibitors: A new class of broad-spectrum dual-targeting antibacterial agents. PLoS ONE, 2013, 8, e84409. [36] Shirude, P.S.; Madhavapeddi, P.; Tucker, J.A.; Murugan, K.; Patil, V.; Basavarajappa, H.; Raichurkar, A.V.; Humnabadkar, V.; Hussein, S.; Sharma, S.; Ramya, V.K.; Narayan, C.B.; Balganesh, T.S.; Sambandamurthy, V.K. Aminopyrazinamides: novel and specific GyrB inhibitors that kill replicating and nonreplicating Mycobacterium tuberculosis. ACS Chem. Biol., 2013, 8, 519523. [37] Liang, X.; Lee, C.J.; Zhao, J.; Toone, E.J.; Zhou, P. Synthesis, structure, and antibiotic activity of aryl-substituted Lpxc inhibitors. J. Med. Chem., 2013, 56, 6954-6966. [38] Koh, C.Y.; Kim, J.E.; Wetzel, A.B.; de van der Schueren, W.J.; Shibata, S.; Ranade, R.M.; Liu, J.; Zhang, Z.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.; Fan, E.; Hol, W.G. Structures of Trypanosoma brucei methionyl-tRNA synthetase with urea-based inhibitors provide guidance for drug design against sleeping sickness. PLoS Negl. Trop. Dis., 2014, 8, e2775. [39] Venkatesan, A.M.; Agarwal, A.; Abe, T.; Ushirogochi, H.; Yamamura, I.; Ado, M.; Tsuyoshi, T.; Dos Santos, O.; Gu, Y.; Sum, F.W.; Li, Z.; Francisco, G.; Lin, Y.I.; Petersen, P.J.; Yang, Y.; Kumagai, T.; Weiss, W.J.; Shlaes, D.M.; Knox, J.R.; Mansour, T.S. Structure-activity relationship of 6-methylidene penems bearing 6,5 bicyclic heterocycles as broad-spectrum β-lactamase inhibitors: 26 Evidence for 1,4-thiazepine intermediates with C7 R stereochemistry by computational methods. J. Med. Chem., 2006, 49, 4623-4637. [40] Knowles, D.J.; Foloppe, N.; Matassova, N.B.; Murchie, A.I. The bacterial ribosome, a promising focus for structure-based drug design. Curr. Opin. Pharmacol., 2002, 2, 501-506. [41] Olivares Pacheco, J.A.; Bernardini, A.; Garcia-Leon, G.; Corona, F.; Sanchez, M.B.; Martinez, J.L. The intrinsic resistome of bacterial pathogens. Frontiers Microbiol., 2013, 4. [42] Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M.; al, e. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 1995, 269, 496-512. [43] Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; Louis, E.J.; Mewes, H.W.; Murakami, Y.; Philippsen, P.; Tettelin, H.; Oliver, S.G. Life with 6000 genes. Science, 1996, 274, 546-567. [44] Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; ColladoVides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; Gregor, J.; Davis, N.W.; Kirkpatrick, H.A.; Goeden, M.A.; Rose, D.J.; Mau, B.; Shao, Y. The complete genome sequence of Escherichia coli K12. Science, 1997, 277, 1453-1462. [45] National Center for Biotechnology Information (NCBI), genome information by organism, http://www.ncbi.nlm.nih.gov/genome/browse/ [46] Chan, P.F.; Macarron, R.; Payne, D.J.; Zalacain, M.; Holmes, D.J. Novel antibacterials: a genomics approach to drug discovery. Curr. Drug Targets - Infect. Disord., 2002, 2, 291-308. [47] Marrakchi, H.; Dewolf, W.E.; Quinn, C.; West, J.; Polizzi, B.J.; So, C.Y.; Holmes, D.J.; Reed, S.L.; Heath, R.J.; Payne, D.J.; Rock, C.O.; Wallis, N.G. Characterization of Streptococcus pneumoniae enoyl-(acyl-carrier protein) reductase (FabK). Biochem. J., 2003, 370, 1055-1062. [48] Zhu, L.; Lin, J.; Ma, J.; Cronan, J.E.; Wang, H. Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother., 2010, 54, 689-698. [49] Kaplan, N; Albert, M; Awrey, D; Bardouniotis, E; Berman, J; Clarke, T; Dorsey, M; Hafkin, B; Ramnauth, J; Romanov, V; Schmid, M.B.; Thalakada, R; Yethon, J; Pauls, H.W. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother., 2012, 56, 5865-5874. [50] Yao, J.; Maxwell, J.B.; Rock, C.O. Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J. Biol. Chem., 2013, 288, 36261-36271. [51] Karlowsky, J.A.; Kaplan, N.; Hafkin, B.; Hoban, D.J.; Zhanel, G.G. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother., 2009, 53, 3544-3548. [52] Gallant, P.; Finn, J.; Keith, D.; Wendler, P. The identification of quality antibacterial drug discovery targets: a case study with aminoacyl-tRNA synthetases. Expert Opin. Ther. Targets, 2000, 4, 1-9. [53] Gentry, D.R.; Ingraham, K.A.; Stanhope, M.J.; Rittenhouse, S.; Jarvest, R.L.; O'Hanlon, P.J.; Brown, J.R.; Holmes, D.J. Variable sensitivity to bacterial methionyl-tRNA synthetase inhibitors reveals subpopulations of Streptococcus pneumoniae with two distinct methionyl-tRNA synthetase genes. Antimicrob. Agents Chemother., 2003, 47, 1784-1789. [54] Brinster, S.; Lamberet, G.; Staels, B.; Trieu-Cuot, P.; Gruss, A.; Poyart, C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature, 2009, 458, 83-86. [55] Wang, J.; Soisson, S.M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y.S.; Cummings, R.; Ha, S.; Dorso, K.; Motyl, M.; Jayasuriya, H.; Ondeyka, J.; Herath, K.; Zhang, C.; Hernandez, L.; Allocco, J.; Basilio, Ã.n.; Tormo, J.R.; Genilloud, O.; Vicente, F.; Pelaez, F.; Colwell, L.; Lee, S.H.; Michael, B.; Felcetto, T.; Gill, C.; Silver, L.L.; Hermes, J.D.; Bartizal, K.; Barrett, J.; Schmatz, D.; Becker, J.W.; Cully, D.; Singh, S.B. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature, 2006, 441, 358361. [56] Wang, J.; Kodali, S.; Lee, S.H.; Galgoci, A.; Painter, R.; Dorso, K.; Racine, F.; Motyl, M.; Hernandez, L.; Tinney, E.; Colletti, S.L.; Herath, K.; Cummings, R.; Salazar, O.; González, I.; Basilio, A.; Vicente, F.; Genilloud, O.; Pelaez, F.; Jayasuriya, H.; Young, K.; Cully, D.F.; Singh, S.B. 27 Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Nat. Acad. Sci., 2007, 104, 7612-7616. [57] Freiberg, C.; Brunner, N.A.; Schiffer, G.; Lampe, T.; Pohlmann, J.; Brands, M.; Raabe, M.; Häbich, D.; Ziegelbauer, K. Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity. J. Biol. Chem., 2004, 279, 26066-26073. [58] von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Antibacterial natural products in medicinal chemistry-exodus or revival? Angew. Chemie Int. Ed., 2006, 45, 5072-5129. [59] Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev., 1997, 23, 3-25. [60] O'Shea, R.; Moser, H.E. Physicochemical properties of antibacterial compounds: implications for drug discovery. J. Med. Chem., 2008, 51, 2871-2878. [61] Wecke, T.; Mascher, T. Antibiotic research in the age of omics: from expression profiles to interspecies communication. J. Antimicrob. Chemother., 2011, 66, 2689-2704. [62] Lima, T.B.; Pinto, M.F.S.; Ribeiro, S.M.; de Lima, L.A.; Viana, J.C.; Júnior, N.G.; Cândido, E.d.S.; Dias, S.C.; Franco, O.L. Bacterial resistance mechanism: what proteomics can elucidate. FASEB J., 2013, 27, 1291-1303. [63] Romero, D.; Traxler, M.F.; López, D.; Kolter, R. Antibiotics as signal molecules. Chem. Rev., 2011, 111, 5492-5505. [64] Gauwerky, K.; Borelli, C.; Korting, H.C. Targeting virulence: A new paradigm for antifungals. Drug Discov. Today, 2009, 14, 214-222. [65] Alksne, L.E.; Projan, S.J. Bacterial virulence as a target for antimicrobial chemotherapy. Curr. Opin. Biotech., 2000, 11, 625-636. [66] Alekshun, M.N.; Levy, S.B. Targeting virulence to prevent infection: to kill or not to kill? Drug Discov. Today: Therap. Strategies, 2004, 1, 483-489. [67] Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol., 2007, 3, 541-548. [68] Barczak, A.K.; Hung, D.T. Productive steps toward an antimicrobial targeting virulence. Curr. Opin. Microbiol., 2009, 12, 490-496. [69] Kandaswamy, K.; Liew, T.H.; Wang, C.Y.; Huston-Warren, E.; Meyer-Hoffert, U.; Hultenby, K.; Schröder, J.M.; Caparon, M.G.; Normark, S.; Henriques-Normark, B.; Hultgren, S.J.; Kline, K.A. Focal targeting by human β-defensin 2 disrupts localized virulence factor assembly sites in Enterococcus faecalis. Proc. Nat. Acad. Sci., 2013, 110, 20230-20235. [70] Rasmussen, T.B.; Bjarnsholt, T.; Skindersoe, M.E.; Hentzer, M.; Kristoffersen, P.; Köte, M.; Nielsen, J.; Eberl, L.; Givskov, M. Screening for quorum-sensing inhibitors (QSI) by use of a novel genetic system, the QSI selector. J. Bacteriol., 2005, 187, 1799-1814. [71] Lowy, I.; Molrine, D.C.; Leav, B.A.; Blair, B.M.; Baxter, R.; Gerding, D.N.; Nichol, G.; Thomas, W.D.; Leney, M.; Sloan, S.; Hay, C.A.; Ambrosino, D.M. Treatment with monoclonal antibodies against Clostridium difficile toxins. New Eng. J. Med., 2010, 362, 197-205. [72] López, E.L.; Contrini, M.M.; Glatstein, E.; González Ayala, S.; Santoro, R.; Allende, D.; Ezcurra, G.; Teplitz, E.; Koyama, T.; Matsumoto, Y.; Sato, H.; Sakai, K.; Hoshide, S.; Komoriya, K.; Morita, T.; Harning, R.; Brookman, S. Safety and pharmacokinetics of urtoxazumab, a humanized monoclonal antibody, against Shiga-like toxin 2 in healthy adults and in pediatric patients infected with Shiga-like toxin-producing Escherichia coli. Antimicrob. Agents Chemother., 2010, 54, 239-243. [73] Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: can we make evolutionproof drugs? Nat. Rev. Microbiol., 2014, 12, 300-308. [74] Gordon, C.P.; Williams, P.; Chan, W.C. Attenuating Staphylococcus aureus virulence gene regulation: A medicinal chemistry perspective. J. Med. Chem., 2013, 56, 1389-1404. [75] Rampioni, G.; Leoni, L.; Williams, P. The art of antibacterial warfare: Deception through interference with quorum sensing–mediated communication. Bioorg. Chem., 2014, 55, 60-68. [76] Antunes, L.C.M.; Ferreira, R.B.R.; Buckner, M.M.C.; Finlay, B.B. Quorum sensing in bacterial virulence. Microbiol., 2010, 156, 2271-2282. [77] Keller, L.; Surette, M.G. Communication in bacteria: an ecological and evolutionary perspective. Nat. Rev. Microbiol., 2006, 4, 249-258. 28 [78] Geske, G.D.; O'Neill, J.C.; Blackwell, H.E. Expanding dialogues: from natural autoinducers to non-natural analogues that modulate quorum sensing in Gram-negative bacteria. Chem. Soc. Rev., 2008, 37, 1432-1447. [79] Zhu, J.; Kaufmann, G.F. Quo vadis quorum quenching? Curr. Opin. Pharmacol., 2013, 13, 688-698. [80] Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol., 2001, 55, 165-199. [81] Pappas, K.M.; Weingart, C.L.; Winans, S.C. Chemical communication in proteobacteria: biochemical and structural studies of signal synthases and receptors required for intercellular signalling. Mol. Microbiol., 2004, 53, 755-769. [82] Gould, T.A.; Watson, W.T.; Choi, K.H.; Schweizer, H.P.; Churchill, M.E.A. Crystallization of Pseudomonas aeruginosa AHL synthase LasI using β-turn crystal engineering. Acta Crystallogr. Sect. D: Biol. Crystallogr., 2004, 60, 518-520. [83] Parsek, M.R.; Val, D.L.; Hanzelka, B.L.; Cronan, J.E.; Greenberg, E.P. Acyl homoserinelactone quorum-sensing signal generation. Proc. Nat. Acad. Sci., 1999, 96, 4360-4365. [84] Chung, J.; Goo, E.; Yu, S.; Choi, O.; Lee, J.; Kim, J.; Kim, H.; Igarashi, J.; Suga, H.; Moon, J.S.; Hwang, I.; Rhee, S. Small-molecule inhibitor binding to an N-acyl-homoserine lactone synthase. Proc. Nat. Acad. Sci., 2011, 108, 12089-12094. [85] Rasmussen, T.B.; Givskov, M. Quorum-sensing inhibitors as anti-pathogenic drugs. Int. J. Med. Microbiol., 2006, 296, 149-161. [86] Eberhard, A.; Widrig, C.; McBath, P.; Schineller, J. Analogs of the autoinducer of bioluminescence in Vibrio fischeri. Arch. Microbiol., 1986, 146, 35-40. [87] Schaefer, A.L.; Hanzelka, B.L.; Eberhard, A.; Greenberg, E.P. Quorum sensing in Vibrio fischeri: probing autoinducer-LuxR interactions with autoinducer analogs. J. Bacteriol., 1996, 178, 2897-2901. [88] Janssens, J.C.A.; Metzger, K.; Daniels, R.; Ptacek, D.; Verhoeven, T.; Habel, L.W.; Vanderleyden, J.; De Vos, D.E.; De Keersmaecker, S.C.J. Synthesis of N-acyl homoserine lactone analogues reveals strong activators of Sdia, the Salmonella enterica Serovar typhimurium luxr homologue. Appl. Environ. Microbiol., 2007, 73, 535-544. [89] Reverchon, S.; Chantegrel, B.; Deshayes, C.; Doutheau, A.; Cotte-Pattat, N. New synthetic analogues of N-acyl homoserine lactones as agonists or antagonists of transcriptional regulators involved in bacterial quorum sensing. Bioorg. Med. Chem. Lett., 2002, 12, 1153-1157. [90] Castang, S.; Chantegrel, B.; Deshayes, C.; Dolmazon, R.; Gouet, P.; Haser, R.; Reverchon, S.; Nasser, W.; Hugouvieux-Cotte-Pattat, N.; Doutheau, A. N-Sulfonyl homoserine lactones as antagonists of bacterial quorum sensing. Bioorg. Med. Chem. Lett., 2004, 14, 5145-5149. [91] Frezza, M.; Castang, S.; Estephane, J.; Soulere, L.; Deshayes, C.; Chantegrel, B.; Nasser, W.; Queneau, Y.; Reverchon, S.; Doutheau, A. Synthesis and biological evaluation of homoserine lactone derived ureas as antagonists of bacterial quorum sensing. Bioorg. Med. Chem., 2006, 14, 4781-4791. [92] Li, M.; Ni, N.; Chou, H.T.; Lu, C.D.; Tai, P.C.; Wang, B. Structure-based discovery and experimental verification of novel AI-2 quorum sensing inhibitors against Vibrio harveyi. ChemMedChem, 2008, 3, 1242-1249. [93] Peng, H.; Cheng, Y.; Ni, N.; Li, M.; Choudhary, G.; Chou, H.T.; Lu, C.D.; Tai, P.C.; Wang, B. Synthesis and evaluation of new antagonists of bacterial quorum sensing in Vibrio harveyi. ChemMedChem, 2009, 4, 1457-1468. [94] Zhu, P.; Peng, H.; Ni, N.; Wang, B.; Li, M. Novel AI-2 quorum sensing inhibitors in Vibrio harveyi identified through structure-based virtual screening. Bioorg. Med. Chem. Lett., 2012, 22, 6413-6417. [95] Schuster, M.; Joseph Sexton, D.; Diggle, S.P.; Peter Greenberg, E. Acyl-homoserine lactone quorum sensing: From evolution to application. Annu. Rev. Microbiol., 2013, 67, 43-63. [96] Dionne, M.S.; Ghori, N.; Schneider, D.S. Drosophila melanogaster is a genetically tractable model host for Mycobacterium marinum. Infect. Immun., 2003, 71, 3540-3550. [97] Jander, G.; Rahme, L.G.; Ausubel, F.M. Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J. Bacteriol., 2000, 182, 3843-3845. [98] D'Argenio, D.A.; Gallagher, L.A.; Berg, C.A.; Manoil, C. Drosophila as a model host for Pseudomonas aeruginosa infection. J. Bacteriol., 2001, 183, 1466-1471. 29 [99] Fuqua, C.; Greenberg, E.P. Listening in on bacteria: acyl-homoserine lactone signalling. Nat. Rev. Mol. Cell Biol., 2002, 3, 685-695. [100] Marra, A. Can virulence factors be viable antibacterial targets? Expert Rev. Anti-infect. Ther., 2004, 2, 61-72. [101] Bjarnsholt, T.; Givskov, M. Quorum-sensing blockade as a strategy for enhancing host defences against bacterial pathogens. Philos. Trans. R. Soc. B: Biol. Sci., 2007, 362, 1213-1222. [102] Waters, C.M.; Bassler, B.L. Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol., 2005, 21, 319-346. [103] Wadhams, G.H.; Armitage, J.P. Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell Biol., 2004, 5, 1024-1037. [104] Stephenson, K.; Hoch, J.A. Virulence- and antibiotic resistance-associated two-component signal transduction systems of Gram-positive pathogenic bacteria as targets for antimicrobial therapy. Pharmacol. Therap., 2002, 93, 293-305. [105] Parkinson, J.S.; Kofoid, E.C. Communication modules in bacterial signaling proteins. Annu. Rev. Genet., 1992, 26, 71-112. [106] Hoch, J.A.; Varughese, K.I. Keeping signals straight in phosphorelay signal transduction. J. Bacteriol., 2001, 183, 4941-4949. [107] Barrett, J.F.; Goldschmidt, R.M.; Lawrence, L.E.; Foleno, B.; Chen, R.; Demers, J.P.; Johnson, S.; Kanojia, R.; Fernandez, J.; Bernstein, J.; Licata, L.; Donetz, A.; Huang, S.; Hlasta, D.J.; Macielag, M.J.; Ohemeng, K.; Frechette, R.; Frosco, M.B.; Klaubert, D.H.; Whiteley, J.M.; Wang, L.; Hoch, J.A. Antibacterial agents that inhibit two-component signal transduction systems. Proc. Nat. Acad Sci., 1998, 95, 5317-5322. [108] Roychoudhury, S.; Collins, S.M.; Hynd, B.A.; Parker, C.N. High throughput autophosphorylation assay for bacterial protein histidine kinases. J. Biomolec. Screening, 1997, 2, 8590. [109] Moellering, R.C. Antibiotic resistance: lessons for the future. Clin. Infect. Dis., 1998, 27, S135-S140. [110] Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: a review. Int. J. Tuberc. Lung Dis., 2003, 7, 6-21. [111] Hu, Y.; Coates, A.R.; Mitchison, D.A. Sterilising action of pyrazinamide in models of dormant and rifampicin-tolerant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis., 2006, 10, 317-322. [112] Hu, Y.; Coates, A.R.M.; Mitchison, D.A. Sterilizing activities of fluoroquinolones against rifampin-tolerant populations of Mycobacterium tuberculosis. Antimicrob. Agents Chemother., 2003, 47, 653-657. [113] Singh, R.; Manjunatha, U.; Boshoff, H.I.M.; Ha, Y.H.; Niyomrattanakit, P.; Ledwidge, R.; Dowd, C.S.; Lee, I.Y.; Kim, P.; Zhang, L.; Kang, S.; Keller, T.H.; Jiricek, J.; Barry, C.E. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science, 2008, 322, 13921395. [114] Ji, B.; Chauffour, A.; Andries, K.; Jarlier, V. Bactericidal activities of R207910 and other newer antimicrobial agents against Mycobacterium leprae in mice. Antimicrob. Agents Chemother., 2006, 50, 1558-1560. [115] Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against Tuberculosis in vitro and in mice. PLoS Med, 2006, 3, e466. [116] Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother., 2005, 56, 968-974. [117] LL-3858. Tuberculosis (Edinburgh, Scotl.), 2008, 88 (2), 126. [118] Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol., 2009, 2, 215-218. [119] Ooi, N.; Miller, K.; Randall, C.; Rhys-Williams, W.; Love, W.; Chopra, I. XF-70 and XF-73, novel antibacterial agents active against slow-growing and non-dividing cultures of Staphylococcus aureus including biofilms. J. Antimicrob. Chemother., 2010, 65, (1), 72-78. 30 [120] Hu, Y.; Shamaei-Tousi, A.; Liu, Y.; Coates, A. A new approach for the discovery of antibiotics by targeting non-multiplying bacteria: A novel topical antibiotic for Staphylococcal infections. PLoS ONE, 2010, 5, e11818. [121] Hu, Y.; Coates, A.R.M. Enhancement by novel anti-methicillin-resistant Staphylococcus aureus compound HT61 of the activity of neomycin, gentamicin, mupirocin and chlorhexidine: in vitro and in vivo studies. J. Antimicrob. Chemother., 2012, 68, (2), 374-384. [122] Cherian, P.T.; Wu, X.; Maddox, M.M.; Singh, A.P.; Lee, R.E.; Hurdle, J.G. Chemical modulation of the biological activity of reutericyclin: a membrane-active antibiotic from Lactobacillus reuteri. Sci. Rep., 2014, 4, 4721. [123] Rami, A.; Toutain, C.M.; Jacq, A. An increased level of alternative sigma factor RpoS partially suppresses drug hypersensitivity associated with inactivation of the multidrug resistance pump AcrAB in Escherichia coli. Res. Microbiol., 2005, 156, 356-360. [124] Murakami, K.; Ono, T.; Viducic, D.; Kayama, S.; Mori, M.; Hirota, K.; Nemoto, K.; Miyake, Y. Role for rpoS gene of Pseudomonas aeruginosa in antibiotic tolerance. FEMS Microbiol. Lett., 2005, 242, 161-167. [125] Pepper, E.D.; Farrell, M.J.; Finkel, S.E. Role of penicillin-binding protein 1b in competitive stationary-phase survival of Escherichia coli. FEMS Microbiol. Lett., 2006, 263, 61-67. [126] Hu, Y.; Coates, A.R.M. Transposon mutagenesis identifies genes which control antimicrobial drug tolerance in stationary-phase Escherichia coli. FEMS Microbiol. Lett., 2005, 243, 117-124. [127] Coates, A.R.M.; Hu, Y. Targeting non-multiplying organisms as a way to develop novel antimicrobials. Trends Pharmacol. Sci., 2008, 29, 143-150. [128] Jefferson, K.K.; Goldmann, D.A.; Pier, G.B. Use of confocal microscopy to analyze the rate of vancomycin penetration through Staphylococcus aureus biofilms. Antimicrob. Agents Chemother., 2005, 49, 2467-2473. [129] Zhang, Y. Persisters, persistent infections and the Yin–Yang model. Emerg. Microbes Infect., 2014, 3, e3. [130] Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol., 2007, 5, 4856. [131] Mitchison, D.A. Antimicrobial therapy of tuberculosis: justification for currently recommended treatment regimens. Semin. Respir. Crit. Care Med., 2004, 25, 307-315. [132] MacGowan, A.; Bowker, K. Developments in PK/PD: optimising efficacy and prevention of resistance. A critical review of PK/PD in in vitro models. Int. J. Antimicrob. Agents, 2002, 19, 291298. [133] Pan, J.; Bahar, A.A.; Syed, H.; Ren, D. Reverting antibiotic tolerance of Pseudomonas aeruginosa PAO1 persister cells by Z-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. PLoS ONE, 2012, 7, e45778. [134] Pan, J.; Xie, X.; Tian, W.; Bahar, A.; Lin, N.; Song, F.; An, J.; Ren, D. (Z)-4-Bromo-5(bromomethylene)-3-methylfuran-2(5H)-one sensitizes Escherichia coli persister cells to antibiotics. Appl. Microbiol. Biotechnol., 2013, 97, 9145-9154. [135] Duckworth, D.H. "Who discovered bacteriophage?". Bacteriol. Rev., 1976, 40, 793-802. [136] Skurnik, M.; Strauch, E. Phage therapy: facts and fiction. Int. J. Med. Microbiol., 2006, 296, 5-14. [137] Borysowski, J.; Weber-Dabrowska, B.; Gorski, A. Bacteriophage endolysins as a novel class of antibacterial agents. Exp. Biol. Med., 2006, 231, 366-377. [138] Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage, 2011, 1, 66-85. [139] Gill, J.J.; Hyman, P. Phage choice, isolation, and preparation for phage therapy. Curr. Pharm. Biotech., 2010, 11, 2-14. [140] Jagusztyn-Krynicka Ek Fau - Wyszynska, A.; Wyszynska, A. The decline of antibiotic eranew approaches for antibacterial drug discovery. Pol. J. Microbiol., 2008, 57, 91-98. [141] Rogers, G.B.; Carroll, M.P.; Bruce, K.D. Enhancing the utility of existing antibiotics by targeting bacterial behaviour? Br. J. Pharmacol., 2012, 165, 845-857. [142] Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotech., 2010, 28, 591-595. 31 [143] Abdul-Hassan, H.; El-Tahan, E.; Massoud, B.; Gomaa, R. Bacteriophage therapy of Pseudomonas burn wound sepsis. Ann. Medit. Burn Club, 1990, 3, 262-264. [144] Jikia, D.; Chkhaidze, N.; Imedashvili, E.; Mgaloblishvili, I.; Tsitlanadze, G.; Katsarava, R.; Glenn Morris, J.; Sulakvelidze, A. The use of a novel biodegradable preparation capable of the sustained release of bacteriophages and ciprofloxacin, in the complex treatment of multidrug-resistant Staphylococcus aureus-infected local radiation injuries caused by exposure to Sr90. Clin. Exp. Dermat., 2005, 30, 23-26. [145] Lazareva, E.B.; Smirnov, S.V.; Khvatov, V.B.; Spiridonova, T.G.; Bitkova, E.E.; Darbeeva, O.S.; Maiskaia, L.M.; Parfeniuk, R.L.; Men'shikov, D.D. Efficacy of bacteriophages in complex treatment of patients with burn wounds. Antibiot. Khimioter., 2001, 46, 10-14. [146] McVay, C.S.; Velásquez, M.; Fralick, J.A. Phage therapy of Pseudomonas aeruginosa infection in a mouse burn wound model. Antimicrob. Agents Chemother., 2007, 51, 1934-1938. [147] Paul, V.D.; Sundarrajan, S.; Rajagopalan, S.S.; Hariharan, S.; Kempashanaiah, N.; Padmanabhan, S.; Sriram, B.; Ramachandran, J. Lysis-deficient phages as novel therapeutic agents for controlling bacterial infection. BMC microbiol., 2011, 11, 195. [148] Abedon, S.T. Phage therapy: Eco-physiological pharmacology. Scientifica, 2014, Article ID 581639, 29 pages, http://dx.doi.org/10.1155/2014/581639. [149] Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol., 2010, 11, 69-86. [150] Verbeken, G.; Pirnay, J.P.; Lavigne, R.; Jennes, S.; De Vos, D.; Casteels, M.; Huys, I. Call for a dedicated European legal framework for bacteriophage therapy. Arch. Immunol. Ther. Exp. (Warsz), 2014, 62, 117-129. [151] Huys, I.; Pirnay, J.P.; Lavigne, R.; Jennes, S.; De Vos, D.; Casteels, M.; Verbeken, G. Paving a regulatory pathway for phage therapy. EMBO reports, 2013, 14, 951-954. FIGURE LEGENDS: Fig. (1). Chemical structure of trimethoprim (1), sulfamethoxazole (2) and a few key inhibitors obtained during SBDD (3-5). Fig. (2). panel A: Flow chart for the process of antibiotic discovery using a genomic approach; panel B: An illustrative example of large screening campaign of GSK. Fig. (3). Representative chemical structures of fatty acid biosynthesis inhibitors. Fig. (4). Representative chemical structures of autoinducer acyl-HSL (10), AIP-1 (11) and AI-2 (12). Fig. (5). Vibrio fischeri LuxI/R quorum sensing circuit (adapted from reference 80). Fig. (6). Chemical structures of natural autoinducer (C8-HSL, 13) and TofI inhibitors. Fig. (7). Representative structures of acyl-HSL antagonists tested against LuxR. Fig. (8). Some examples of AI-2 mediated QS (LuxPQ, the receptor protein) inhibitors. Fig. (9). Basic two-component signal transduction system (TCSTS) (adapted from reference 104). 32 Fig. (10). Representative structures of different chemical classes as TCSTS inhibitors. Fig. (11). Conventional treatment of infections: multiplying bacteria die when treated with antibiotics (left panel) while little or no effect is noticed on non-multiplying bacteria (right panel). Non-multiplying bacteria can serve as a reservoir for multiplying bacteria and are responsible for recurrence of the infection and emergence of resistance (adapted from reference 20). Fig. (12). Existing antibiotics effective in killing non-multiplying bacteria. Fig. (13). New drug candidates in clinical trials which target non-multiplying bacteria. FIGURES 33 Fig. (1). Chemical structure of trimethoprim (1), sulfamethoxazole (2) and a few key inhibitors obtained during SBDD (3-5). 34 Fig. (2). panel A: Flow chart for the process of antibiotic discovery using a genomic approach; panel B: An illustrative example of large screening campaign of GSK. 35 Fig. (3). Representative chemical structures of fatty acid biosynthesis inhibitors. Fig. (4). Representative chemical structures of autoinducer acyl-HSL (10), AIP-1 (11) and AI-2 (12). Fig. (5). Vibrio fischeri LuxI/R quorum sensing circuit (adapted from reference 80). 36 O O O N H N O O O N N H OH C8-HSL (13) J8-C8 (14) E9C-3oxoC6 (15 ) Fig. (6). Chemical structures of natural autoinducer (C8-HSL, 13) and TofI inhibitors. Fig. (7). Representative structures of acyl-HSL antagonists tested against LuxR. 37 Fig. (8). Some examples of AI-2 mediated QS (LuxPQ, the receptor protein) inhibitors. 38 Fig. (9). Basic two-component signal transduction system (TCSTS) (adapted from reference 104). 39 Fig. (10). Representative structures of different chemical classes as TCSTS inhibitors. 40 Fig. (11). Conventional treatment of infections: multiplying bacteria die when treated with antibiotics (left panel) while little or no effect is noticed on non-multiplying bacteria (right panel). Non-multiplying bacteria can serve as a reservoir for multiplying bacteria and are responsible for recurrence of the infection and emergence of resistance (adapted from reference 20). Fig. (12). Existing antibiotics effective in killing non-multiplying bacteria. 41 Fig. (13). New drug candidates in clinical trials which target non-multiplying bacteria. 42