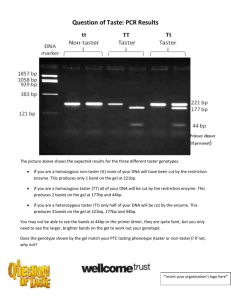
Dinman Lab Protocols
advertisement

Dinman Lab Protocols Dinman Lab Protocols ............................................................................................................. 1 Ethanol Precipitation ............................................................................................................... 3 Smash and Grab ...................................................................................................................... 4 Smash and Grab (cheater version) ......................................................................................... 5 Estimating Nucleic Acid concentration via Optical Density ................................................. 6 Getting plasmid or yeast stocks from the -80 freezer. .......................................................... 7 Preparing stocks for the -80 freezer. ...................................................................................... 7 Performing a plasmid miniprep............................................................................................... 8 Cutting and isolating fragments from plasmids (gel purification) ........................................ 9 qRT-PCR of Bglobin from Mammalian Cells (Vero) ..............................................................10 Restriction Digestions ............................................................................................................17 Maintenance of HeLa cells. ....................................................................................................21 Trey’s current Transfection protocols ...................................................................................24 Dual luciferase with the Simon Lab Luminometer ................................................................26 Flow Cytometry and Fluorescence Activated Cell Sorting ........ Error! Bookmark not defined. Alternate FACS cell staining method (Song lab) ..................................................................34 Western Blot ............................................................................................................................36 Bacterial Transformation ........................................................................................................41 Yeast Transformation .............................................................................................................42 Cell culture ..............................................................................................................................43 Plasmid Construction .............................................................................................................45 -1 PRF quantification by dual luciferase ...............................................................................47 -1 PRF quantification by in-vitro transcription......................................................................48 qPCR ........................................................................................................................................49 Mammalian time-course assays.............................................................................................50 RNAi .........................................................................................................................................51 Affinity purification .................................................................................................................52 Electrophoretic Mobility Shift Assays ...................................................................................53 Chemical Protection Assays ..................................................................................................54 In-vitro transcription for molecular tweezers ........................................................................55 Common Lab Buffers..............................................................................................................56 1 References ..............................................................................................................................57 2 Ethanol Precipitation 1. Ethanol precipitation is the most common methodology used in the lab. It is a simple and fast way to isolate nucleic acids away from surrounding media, to concentrate nucleic acids, and ready nucleic acids for storage in an appropriate buffer. 2. Simplest: Add 2.5 volumes of ethanol, freeze overnight at -20C, spin down 5-10 minutes in the cold room, dry pellet, resuspend. a. Salt helps nucleic acids come out of solution. Thus we often use 3M NaOAc (pH 5.2)Add 0.1 volumes of NaOAc along with the 2.5 volumes of ethanol. b. Unless you are using soap agents (SDS), 3M KOAc may prove better. c. RNA especially prefers a lower pH, thus it is important to ensure pH 5.2 of your salt solution. 3. Salts can contaminate enzymatic reactions and lead to very strange looking agarose gels. a. After precipitation in the -20C, remove the ethanol and rinse the pellet with 2.5 volumes of 70% ethanol. Mix sufficiently to break the pellet. b. Refreeze at -20C for >= 1 hour or -80C for >= 15 minutes. c. Spin down again. The resulting pellet will be less visible and stick less to the tube, so care must be taken when removing the ethanol. 4. After removing ethanol, dry the remaining pellet. Most protocols warn against using a vacuum drier. This is because over-drying results in nucleic acids which are much less soluble. 5. Elution may be done in a few buffers. H2O, TE, an SSC storage buffer (RNA) a. 10mM SSC, HCL: 0.0294g SSC in 10ml H2O + 3.5ul 10M HCl. 6. Low abundance precipitations benefit from an additional seed agent, usually glycogen a. Add 1-2ul of good glycogen (Roche on the door of the -20) 3 Smash and Grab We use a slightly modified version of the “Bust n’ Grab” protocol.(Harju, Fedosyuk, and Peterson 8) which in turn came from the “Smash and Grab protocol” (Hoffman Unit13) Here is the short version: 1. Grow an overnight culture of yeast. 2. Spin down cells (1200g -- slow), remove media and resuspend in 500ul water. 3. Transfer to microcentrifuge tube and spin down briefly. (Or spin down multiple times directly from culture tubes to microcentrifuge) 4. Resuspend cells in 200ul smash and grab buffer, add 0.3g glass beads (~ 200ul volume) a. Add 200ul Phenol Chloroform. 5. Vortex at maximum speed 3 minutes. 6. Add 200ul TE buffer and vortex briefly. 7. Centrifuge maximum speed 5 minutes. 8. Remove aqueous layer to new tube, add 1ml 100% EtOH, mix. 9. Precipitation should be nearly instantaneous, if so: a. Spin down 3 minutes at RT, full speed. Dry the pellet and resuspend in TE buffer. 10. Add 30ul of 1mg/ml DNase-free RNase, mix and incubate 5-10 minutes @ 37C. 11. Ethanol precipitate and resuspend in TE buffer. Smash and Grab buffer: (from protocol) 1. 2% triton-X 100 2. 1% SDS 3. 100mM NaCl 4. 10mM Tris:Cl pH 8.0 5. 1mM EDTA pH 8.0 My Smash and Grab buffer 1. 100ml 10% SDS 2. 20ml 100% Triton-X 100 3. 20ml 5M NaCL 4. 10ml 1M Tris pH 8.0 5. 2ml 0.5M EDTA 6. 350ml H2O 4 Smash and Grab (cheater version) 1. Grow 5ml cultures overnight. 2. Remove 1.0-1.6ml aliquots into eppendorf tubes and spin down 1m @ 2000rpm a. Repeat until all cells are used 3. Add 400ul smash and grab buffer (above) 4. Vortex, add beads (100-500ul) 5. Add 500ul Phenol Chloroform (higher pH is better for DNA, low pH for RNA) 6. Vortex 2 min in the cold room, spin down max speed 4 min in the cold room. 7. Transfer upper phase to a new tube (Usually 10-50ul is lost per transfer) a. I usually have these tubes pre-aliquoted with Phenol 8. Re-extract in phenol chloroform 1-3 times (usually twice) a. For repeats, 1 minute vortexes is fine. 9. Optionally extract with 400ul pure chloroform. (to dilute out any remaining phenol.) 10. Ethanol precipitate 11. Elute in 40-100ul H2O or TE. 5 Estimating Nucleic Acid concentration via Optical Density Optical Density measurements of nucleic acids take advantage of Beer’s Law: Absorbance = concentration * path length * extinction coefficient The average ext. coefficient for DNA is 0.02 ml/ug*cm and RNA is 0.027 ml/ug*cm (Interestingly, short DNA molecules differ based on length and base composition -check the IDT information sheets for our oligonucleotides if you don’t believe me) Therefore we can take a shortcut and assume the following: 1. 1ul of a DNA sample with an OD of 1.0 has 50ng of DNA. 2. 1ul of a total cellular RNA sample with an OD of 1.0 has 40ng of RNA. 3. A ‘good’ sample of nucleic acid will have a ratio of OD260/OD280 of 1.8<=x<=2.0 a. Ratios significantly below 1.8 suggest protein contamination (As I understand it, OD280 catches the ring containing amino acids (especially tryptophan, but also tyrosine and phenylalanine to some extent)). Also phenol contamination might decrease this ratio. i. If these nucleic acids came from a dirty prep (phenol chloroform) then wash some more. b. Ratios significantly higher than 2.0 generally suggest salt contamination. i. Do a better rinse with 70% ethanol during your ethanol precipitation. Using our Spectrophotometer 1. Use only cuvettes which are certified for UV light or the very expensive quartz cuvettes. 2. Load a dilution of water + nucleic acid 3. Use reading mode #4 on the spectrophotometer in order to read 260nm and 280nm and print the results. 4. Load a water control cuvette in the bottom well of the spec 5. Load samples above 6. Read and print each result. 7. Calculate the concentration and note the quality. a. I usually have 1ml of water and add 5ul of sample, so I just multiply the OD * 10050 to get ng/ul or just 10,000 to get an estimate. Using the nanodrop in the Song lab 1. Wash the pedestal 2. Load 1ul, hit read, write down result, wash pedestal. 6 Getting plasmid or yeast stocks from the -80 freezer. 1. Figure out the appropriate plasmid(s) using the plasmid database on the main lab computer and/or ApE. 2. Get correct plates from the cold room (LB+carb for bacteria, YPAD or H- for yeast) and let them warm up for a moment, Get some ice to put the cells on while they are out of the -80, get a Bunsen burner running next to your plates. 3. Go to the -80 freezer in 2136 and select the correct box(pJD for plasmids, YJD for yeast strains), take out the vials with the ones you need, put them on ice. a. Try to be relatively quick, each full freeze/thaw cycle of bacteria/yeast kills approximately 50% of the cells in the tube. 4. Using a stick, acquire a dab of frozen cells and streak it out on the plate. 5. Put the plate in the correct incubator for your purpose (usually but not always: 37C for bacteria, 30C for yeast) 6. Allow to grow overnight for bacteria, sometimes longer for yeast, pick a single colony and inoculate the appropriate liquid media If you don’t mind poor quality, it is possible though generally frowned upon to take the stock directly from the -80 and place it into liquid media to grow overnight. Preparing stocks for the -80 freezer. 1. Create a 5ml overnight growth stock of the yeast/bacteria in the appropriate selective media. 2. Have an aliquot of 30% glycerol ready. Glycerol is insanely viscous, so be ready. 3. Document the stocks in the appropriate database on the main computer, the plasmid book, the strain document, etc… 4. Spin down the cells (be gentle with yeast! I never do more than 2,000 rpm) 5. Resuspend the cells in 15% glycerol, I usually suck out all the media, add 800ul of fresh media and 800ul of 30% glycerol. 6. Place the resuspended cells/glycerol in a properly labeled cryogenic tube and into the appropriate box in the -80. 7 Performing a plasmid miniprep We do most of our minipreps using the fermentas kit or something similar, they all follow the same basic pattern and are all derivatives of the alkaline extraction method(Birnboim 243-55) 1. Grow an overnight culture 2. Spin down 5 minutes at full speed, throw away aqueous 3. Add 250ul of the lysis buffer containing RNAse (don’t use this pipette for RNA work, it will be forever contaminated), mix by inversion. 4. Add 250ul of the NaOH buffer, inversion mix. You should have a stringy goopy mess now, the stringy bits are actually DNA 5. Add 350ul of the Acetic Acid buffer, inversion mix. You should have a white precipitate now. 6. Spin down 5-10 minutes at full speed. 7. Load aqueous on a silica binding column 8. Spin down 1-2 minutes, throw away aqueous 9. Wash 1-2 times with the ethanol washing buffer, spin down, throw away aqueous. 10. Spin down one last time to dry the column 11. Add 20-60ul (usually 50ul) elution buffer or water, let sit a couple minutes and spin down into a properly labeled eppendorf tube. 12. Yields vary from 50-500ng/ul, quantify it if you need to know. 8 Cutting and isolating fragments from plasmids (gel purification) The following instructions are a short-cut version taken from the manuals we have in the lab, see those for more detail. 1. Follow the manufacturers instructions for performing the restriction digest http://www.fermentas.com/techinfo/re/index.html a. This usually entails a 10x buffer and one or more enzymes b. Aliquot DNA (approximately 1ug), water, buffer, and enzyme (usually 1ul, 510 units) c. Mix gently, spin down gently d. Allow to sit at 37C for 1-16 hours. 2. Run the resulting DNA fragments out on an approximately 1% agarose gel a. Caveats: The agarose and ethidium bromide from this process are bad for most downstream applications, the gel purification process tries to get around these problems. 3. Excise the fragments using a razor blade and place in a fresh labeled tube. a. Caveat: The UV light is moderately hazardous to us, and very much bad for the DNA sample, don’t take too long. 4. Weigh the gel/nucleic acid sample, the average tube weights 1.04 grams. 5. Add 1,000ul of binding buffer for 1.0g of agarose, place on a 55-60C heating block for 5-10 minutes until the agarose has dissolved. 6. Place the dissolved agarose on a binding column, spin down (1-2 min at max), throw away aqueous. 7. Add another aliquot (usually 400ul) of binding buffer, spin down, throw away aqueous. 8. Add approximately 700ul of wash buffer to the column, spin down and throw away 9. Repeat (I sometimes skip this) 10. Dry the column with another 1-2 minute spin 11. Elute in buffer or water. I have found that traces of the buffers remain after this process and lead to difficulties in taking an OD 260/280 measurement as well as lowered ligation efficiency, but doing the second ethanol rinse step loses a lot of my nucleic acid. So I often instead do an ethanol precipitation after finishing this and take an OD260/280 after the precipitation. 9 qRT-PCR of Bglobin from Mammalian Cells (Vero) SDM of Brewer plasmids 1. Brewer plasmids are described in “Methods in Enzymology Volume 449” found in ‘Vitalsource’ on seenoevil a. Bglobin wt (pJD976) : Full length Bglobin with introns and UTRs b. Bglobin + ARE in 3’UTR (pJD975) : Same with ARE c. pTET Off (pJD979) : Encodes the transactivator protein to turn Bglobin on 2. The CCR5 fragments inserted into pJD975/976 are found in: a. pJD827 (wt), pJD844 (ssd), pJD845, 846, 854-858 3. The SDM was performed as a two stage “mega-oligo” a. Amplify fragments from pJD827 etc with 5’ and 3’ ends corresponding to Bglobin b. Amplify full length insertion plasmids using these 600+bp fragments and pJD975/976 c. Resulting in pJD973-979 Oligos for amplifying CCR5 from pJd827etc look like: Ggtgcatctgtccagtgaggagaagtctgccgttgagcgagttctcaaaaatgaac (57% GC, 67C Tm) (42% GC, 59C Tm) Orange identical to 32bp into exon1 of the Bglobin gene, purple corresponds to the dual luciferase Renilla sequence from any of our dual luciferase plasmids Ccttgccccacagggcagtgaccgtttttggcgtcttcgagctca (70% GC, 68C Tm) (50% GC, 60C Tm) Orange corresponds to the reverse complement of Bglobin exon 1 starting at position 33, purple corresponds to the reverse complement of the Firefly sequence from any dual luciferase plasmid In parentheses next to each oligo are their GC content and melting temperatures with respect to the dual luciferase plasmid and Bglobin plasmid in orange and purple respectively. 4. A 40 cycle PCR reaction was performed using Taq polymerase, these oligos, and pJD827 etc in order to amplify the bglobin end containing dual luciferase plasmid/CCR5 fragments 5. Once conditions were worked out for maximal amplification (different for each plasmid background), plasmid product was gel excised/purified/ethanol precipitated and quantified by OD260/280. It turns out that doing the OD260/280 was not necessary, because all of the PCR product was used in the SDM step. 6. Initially, default SDM conditions were chosen for performing the mega oligo SDM (Stratagene), those did not work. Many attempts were made and success occurred completely randomly. It appears that lowering the Tm, lowering the extension temperature, increasing the extension time, and increasing the number of cycles helped. Unfortunately I do not have the exact conditions which worked, because I tried many different conditions at the same time and never found a trend. 10 7. Sending these plasmids for sequencing must be done multiple times, there are multiple strange crossover events which lead to apparent positive colonies which in fact have indel mutations, tandom duplications, etc… 8. A very few plasmids result which have Bglobin which looks like: New DNA from 1 to 1437 Rabbit_bglobin_exon1Rabbit Bglobin intron1 (1...32) (387...512) Rabbit_Bglobin_exon1_continued (327...386) Rabbit bglobin intron 2 (736...1308) Rabbit bglobin exon 3 (1309...1437) Rabbit bglobin exon 2 (513...735) CCR5 insertion with Dual luciferase ends (33...326) Growing Vero cells for transfection 1. Cells were removed from the -80 at passage 32 from Ewan Plant’s stocks (non ideal) 2. Initial wake up consisted of laying out 15ml of DMEM+FBS in a t75 plate prewarmed to 37C, removing a vial of Vero cells from the -80 and placing it into 37C water for ~5 minutes, then pipetting its contents directly into the media containing T75 and incubating for 3 days. a. The incubator must maintain 5% CO2, 37C, and a humid atmosphere. 3. Each day the flask was removed for a moment to check for contamination and growth state of the cells. 4. When cells reach 80-90% confluency they were split to either another T75 or 6 well plate for transfections. (we usually added 250ul cells to a new T75 and 1-2ml of cells to each well of the transfection plate) 5. Wait another day before transfections. Triple transfections of Bglobin plasmids into Vero cells. 1. pJD175f was chosen as the source of our reference gene(s). This provides two easy oligo pairs, one to amplify Renilla, one for Firefly. Most analyses so far have focused on the 5’ gene (Renilla). 2. We were also given a GFP plasmid by the Song lab (pJD1033) but have not used it very often, citing some papers which suggest that GFP can lead to difficulties for mammalian cells harboring it, though it appears that Vero cells are not susceptible to these problems. 3. pJD979 must accompany any and all Bglobin plasmids used in these assays. It encodes the tTA transactivator protein which is required for expression of Bglobin under the control of the pCMV early promoter (in other words, the Bglobin encoded on pJD973-977). This promoter is activated by the C-terminal activation domain of the VP16 protein of herpes simplex and without it, no Bglobin is expressed. (Resnitzky et al. 1669-79) 11 4. pJD973-pJD978 finally our constructs of interest, they must be endotoxin-free prepared and nanodropped. 5. All transfections followed the instructions of the Fugene 6 protocol. We started out trying 3:2, 3:1, 6:1, and 8:1 reagent:DNA ratios, but eventually settled on 3:1 and 6:1 as the best. (3:1 is the best of all) 6. The best results were found when we mixed the 3 plasmids together in one tube, laid out the 3x quantities of Fugene+media in another tube, then mixed, incubated, and poured onto the cells as described in the protocol. 7. Wait 24-36 hours before isolating mRNA. mRNA isolation of transfected cells. 1. In every instance so far we have had to wait > 1 day before isolating mRNA. As a result, most transfections occurred at the end of ‘Day 1’ and mRNA isolations began at the beginning of ‘Day 3’. 2. Before isolation, cells were trypsinized in the 6 well plates and the cell scrapers were NOT used to remove cells from the plates into eppendorf tubes. The cells were then gently centrifuged and transferred into 100ul fresh DMEM without FBS. 3. At this point we followed the Ambion RNAqueous kit instructions, the only caveat being that for some samples it was necessary to spin the mRNA down for longer than the recommended 10 seconds in order to get all the lysate to spin through the column (the cellular material slightly clogged it). 4. We did however allow the DNAse to incubate for longer than the proscribed 10 minutes, but it would appear later that more time would have been better. 5. All mRNA samples were immediately nano dropped. cDNA synthesis 1. cDNA synthesis followed exactly the protocol in the iScript cDNA synthesis kit 2. 500ng of each sample was used to make cDNA in an attempt to lower the variability between samples of the extension curves in the actual qRT-PCR runs. Performing the actual qRT-PCR The actual qRT-PCR reactions were performed in a mix of the Brewer protocol and the default conditions listed for the Roche LightCycler 480 enzyme mix instructions 1. All cDNAs were diluted to 1:10 for all experiments, The standard curves suggested that we could go as low as 1:10,000 but any further would lead to too much error. 2. Oligos were usually added to 10pMol / reaction, usually diluting some stock 100pMol/ul to 1:20 and adding 2ul / well. 3. The number of wells for each oligo pair was over-estimated by 2-10 wells in order to allow for some No Template Control wells as well as some mRNA negative control wells. 4. Thus, for an experiment of 3 replicates comparing 4 3:1 transfection mRNA samples we might make two master mixes assuming 20 samples each (8 additional samples for the NTCs and mRNA controls), these master tubes would contain: (see 7f for more detail) 12 a. 40ul of 5’ oligo (either reference or target, diluted 1:20 from stock) b. 40ul of 3’ oligo (same) c. 200ul 2x SYBR green master mix (kept in the dark) d. 80ul H2O 5. 18ul of this mix would then be aliquoted per well for our plate, generally splitting the plate in ½, one side for the target gene (bglobin) and one for the reference (renilla); negative controls would be set beside the relevant samples. a. The plate may be left overnight at 4C. b. The thermocycler conditions are as follows: i. 10s @ RT (to get the machine started) ii. 5m @ 95C (this activates the polymerase, which has a bound blocking agent) iii. 10s @ 50C (This corresponds to the Tm of the Brewer Bglobin oligos) iv. 20S @ 72C (extension is really fast, the PCR product is only 90200bp), measure SYBR green during this step 1. Repeat 2-4 50 times v. Perform Tm analysis (constant measuring while slowly increasing temperature from RT -> 95C) Working with the Roche Software This section will deal with using the Roche 480 thermocycler’s software both to work with the qRT-PCR plate and to analyze the data provided. 1. Logging in: Username: dinman Password: Drevil1 2. This brings up the ‘Navigator’ view. When starting up a new plate, I go to the option labeled ‘Open a new experiment using a template’, this opens a new box with 3 panels, inside the ‘protocol’ panel, I scroll down until I find the Dinman protocols. The previous 50 cycle, 50C Tm protocol is found there as ‘Dinman Bglobin…’ Clicking on this brings you to a view of the plate and a ‘start’ button. Click it, give a name for the new experiment, and leave. 3. After the run has finished, its data is automatically saved to the Dinman/Experiments directory under the name chosen in step 2. When you return to the machine, someone else may have logged in and/or turned the software off. Assuming this, log in, bring up the navigator view and click on ‘Open Object.’ This brings up the filesystem view, go to Dinman/Experiments and find the experiment of interest. 4. Clicking on that brings up a schematic of the plate, clicking on individual wells of the plate will show the amplification curve data for each well in a window at the bottom right. 5. IMPORTANT Exporting data from this software is non-trivial, BUT THERE IS A RELATIVELY EASY WAY. Right click on any chart or table in the software and click ‘export chart/table’ 1. This in turn opens up a two panel dialog box which may be used to save a picture of a chart and/or a text file containing the data making up the associated picture. The text files so generated are tab separated and so may be easily opened in Excel a. For example, here is a graph created by taking the raw curves of a Bglobin plate, exporting them to excel, and graphing them: 13 2. The first task when dealing with a new plate is to label all the wells of the plate. This is done via the ‘Sample editor’ It cannot be easily described until you actually try it, but the key points are: 1) First select all of the reference samples on the plate and give them a single name associated with their oligo in the bottom panel and click on ‘reference’, this will turn their wells gray on the picture of the plate above. 2) Select all the target samples (in this case Bglobin), give them another name and click on ‘target’ at the bottom, thus turning them light blue. 3) Then select each set of replicates by sample type (depending on how the plate was laid out, you can do this by clicking on the letter of the row of the plate), give them a name corresponding to the amplicon created from them (wt/ssd/ccr5/etc) in the middle panel and click ‘Make replicates’ at the bottom of the screen. 4) Continue until all samples are allocated, including setting up the NTC and mRNA controls. 3. Once this is complete, I perform my first analysis, the Tm genotype for every well of the plate. Below is an example, showing the Tms for good 90bp Bglobin products (blue), good 200ish bp Renilla products (Red) and a few bad wells. 4. Write down the well numbers of any wackjob wells. They will be shortly excluded from further analyses. Then go back to the initial amplification curve graphs shown when you first opened up the experiment. Clicking on each group of wells as you defined them in the Sample view should bring up a series of amplification curves which all come up at pretty much the exact same time. Ideally the only exceptions will be those few wackjobs identified by the Tm genotyping, but this will not always be the case (below is an example, where I used excel to graph a set of 5 amplifications of Bglobin of which one is an obvious outlier with the same Tm), the point being, identify these wells and write them down too. 14 45 40 35 30 25 20 15 10 5 0 -5 0 10 20 30 40 50 60 5. Once all bad wells have been identified, I go to the ‘Subset’ editor and make an exclusion subset. This brings up another plate view with the default subset of everything listed. Click on the + sign, select every well, then go back and shift-select the wells to be excluded. When finished, the picture of the plate should be mostly blue with the excluded wells shown as inset-white. I note this because it is difficult to tell the difference between a plate where all but 3 wells are included and one where only 3 wells are included. 6. Once bad wells have been excluded, we can make some real analyses. This actually takes very little time, just hit the ‘analysis’ button, click ‘Advanced Relative,’ the 2nd item of this dialog box asks for which subset to use, change it from the default of everything to your exclusion subset, and hit ok. This will bring up another view of the plate with the associations of reference/target wells listed, hit the ‘calculate’ button. A graph should appear which shows the relative ratios of target:reference gene in every group of wells on the plate. 7. As you can see, the graphs are really ugly, but can be quickly exported by right clicking. In addition, the numbers which make them up may be exported by doing the same thing, clicking ‘data’, and bringing them to excel/prism/whatever to be massaged. 15 Target/Reference Ratio 45 40 35 30 25 20 15 10 5 0 8. 16 Restriction Digestions Taken online from molecularstation.com from Maniatis Restriction Enzyme Digestion Restriction Enzymes Restriction Digestion Table of Contents * Restriction Digestion Introduction * Restriction Enzyme Digestion Protocol * Restriction Enzyme Digestion Protocols * Restriction Digestion Videos * Restriction Enzyme Digestion Forum * Restriction Enzyme Digestion Help * Restriction Digestion Troubleshooting ©2006 Molecular Station. Updated July 2008. Information on how to become an expert on Restriction Enzymes and digesting DNA ! Background on Restriction Enzymes Restriction enzymes (or restriction endonucleases) are enzymes that cut doublestranded DNA by making two breaks, one through each of the phosphate backbones of the double helix. The enzyme does this without damaging the bases of the DNA. Although the enzyme breaks the DNA, the chemical bonds can be reformed by other enzymes known as DNA ligases. Therefore, restriction fragments of DNA from different chromosomes or genes can be ligated together, providing their ends are complementary. Usefulness of Restriction Enzymes in Molecular Biology The fact that DNA could be manipulated and different DNA pieces could now be spliced together, opened up new areas of research never before imagined. Many of the methods in molecular biology today rely on restriction enzymes. "Restriction" is derived from the fact that these enzymes were initally discovered in E. coli which appeared to be restricting the infection of specific bacteriophages ("viruses" of bacteria). It is believed restriction enzymes are a host defense mechanism evolved by bacteria and other organisms to resist viral infections, and to help with the elimination of viral sequences. In 1978, the Nobel Prize for Medicine was awarded to Werner Arber, Daniel Nathans and Hamilton Smith for their discovery of restriction endonucleases, which lead to the development of recombinant DNA technologies. The first practical use of restriction enzymes in science and medicine was the manipulation of E. coli bacteria to express recombinant human insulin for diabetics. 17 Handling and Using Restriction Enzymes A unit of restriction enzyme activity is defined by the amount of restrition enzyme required to cut 1 microgram of bacteriophage lambda DNA to completion in a time of 1 hour. Assays developed by the manufacturer of the enzymes are most likely done using highly purified DNA (i. e. plasmids or lambda phage DNA) as substrate, and assay conditions that produce the best results with their particular preparation. Often the laboratory conditions are not as ideal, and a slight excess of enzyme or a longer incubation period is used to help ensure complete digestion. There are many 'universal' enzyme buffers which will work with a variety of enzymes, but often they do not meet the most efficient requirements for any one enzyme. Because the universal buffers are not as efficient, we do not recommend their routine use in the lab (especially for complex genomic DNAs; digests of complex genomic DNAs are usually at high DNA concentrations which will inhibit the enzyme; also, the relatively crude DNA preps may contain inhibitors which requires more units enzyme ard longer incubation times for complete digestion). It is always best to use the manufacturer's recommended assay conditions for restricition digestion. Some manufacturers have cloned enzymes which have very different requirements from other versions of the same enzyme, so check before using. Enzyme concentrations are always given on the side of the enzyme tube. Restriction enzymes used in the lab are always stored at -20 degrees C (in a glycerol base), and should be kept as close to -20 degrees C as possible to extend the life of the enzyme. When setting up digests, bring the reaction tube to the enzyme freezer in an ice bucket; remove the enzyme tube from the freezer and keep the tube on ice while working with the enzyme. Immediately return the tube to the freezer when finished. Use the pipetmen designated for restriction enzymes only and always use a fresh pipetman tip when removing enzyme from the stock tube. Tips on Restriction Enzyme Usage * All manufacturer's of restriction enzymes supply specific buffers with the enzymes, and these should be stored in the -20°C freezer. Restriction Enzymes should also be stored at -20°C. * Never make up restriction digests with the restriction enzyme composing more than 1/10 of the final volume. This is due to the fact that the restriction enzyme proteins are stored in glycerol. At concentrations above 10%, glycerol not only inhibits the digestion but also can cause star activity - leading to aberrant, non-specific cuts of the DNA. * Set up a control digest when using restriction enzymes. This helps you know if the enzyme is working properly and you set the reaction up well. Use a plasmid DNA which generates known fragmentation patterns. * When preparing double enzyme digests, determine the salt content of the buffers. Conduct restriction digestion using the enzyme with the lower salt requirements FIRST, 18 then use the second enzyme by adjusting the reaction tube's buffer to the second optimal salt concentration. * DNA preparations may have impurities which can inhibit restriction enzyme digestion activity. Some DNA isolation kits even use high EDTA buffers to elute the DNA. This is ok for DNA storage, but high concentrations of EDTA can inhibit restriction digestion. PCR purify your DNA and elute with TE or water if your enzyme activity is inhibited but your DNA looks ok. You could also add Mg2+ to the reaction and see if that helps also. * If you cannot obtain complete restriction digestion of your DNA after adding extra enzyme, set up a new digest and add spermidine to a final concentration of 2 mM. Restriction Enzyme Digestion of DNA Analytical versus Preparative Restriction Enzyme Digestions Usually DNA digestions are conducted in 25 - 50 ul (microliters), when you just want to check or analyze your DNA. These are called analytical restriction enzyme digestions. One can digest 0.25 to 2 ug (micrograms) for analytical preparations. This is because on an agarose minigel (30ml), one can visualize approximately .05ug (50 ng, nanograms) per band. Visualization of a band depends on the length of the DNA and the amount of DNA present. In general, the longer the DNA present in the band, the easier is will be to see. DNA digestions of many micrograms and higher volumes from 50 - 500 ul (microliters) or more are preparative, meaning that you are preparing DNA fragments for subsequent purification and cloning. Restriction Digestion of DNA protocol Recipe for Restriction Enzyme Digestion: Analytical 10X Restriction Enzyme Buffer 10 ul DNA (many micrograms of DNA) "X" ul DNA 20 or more ul Water dH2O(millipore or autoclaved) 86 - X ul Restriction Enzyme (10-20 U) 4 ul Total 100 ul You might add Mg2+ to the digestion reaction (in the form of MgCl) if you use a large volume of DNA. You should do this especially if the buffer you eluted your DNA in contained EDTA. This is because if "X" is large, ie > 20ul volume, you may need to add Mg. EDTA binds to 4 moles of Mg for every mole of EDTA. Thus, DNA in a 1 mM 19 (milimolar) solution of EDTA will bind 4mM of Mg. A good rule of thumb is to add 1 1ul of 10mM MgCl to every 20ul of DNA. Preparative 10X Restriction Enzyme Buffer 5 ul DNA "X" ul DNA 0.25 to 10 ul Water dH2O(millipore or autoclaved) 43 - X ul Restriction Enzyme (10-20 U) 1 ul Total 50 ul Incubate at 37°C for 1 - 3 hours. 1 unit is enough enzyme to digest1 ug DNA at least in 1 hour. Digesting for 3 hours you could digest higher amounts of DNA. As you are adding 10-20 Units of enzyme, you could even digest 10ug of DNA in 1 hour ! Enzymes are expensive don't waste them needlessly! Notes on Restriction Enzyme Usage : * Keep enzymes cold. * Minimize the time you have restriction enzymes on ice. You can do this easily by purchasing a cold box (which is basically a block of metal you keep your enzymes in) 20 Maintenance of HeLa cells. This was a gift from Sergey’s previous lab. Go to the bottom to see my new version of maintaining media stocks. Quick caveat: CHO cells like MEMα, not DMEM. A schedule of cell maintenance, feeding, and passaging should be developed to maintain appropriate cell density, nutrient concentration, and pH levels in cell cultures. Cells are best passaged when they are growing exponentially, at 70-80% confluency. This is an example of the schedule used for routine maintenance of HeLa cells. Cells are maintained on Mondays and Fridays, but any combination of 3 nights/4 nights schedule is suitable. Also adjust this schedule as observations are made regarding cell growth. Volumes are given for a T25 flask and so need to be adjusted for T75 flasks. Avoid trypsinizing the cells you are carrying forward too often. This selects for trypsin resistant cells and causes the passage count to increase too quickly. Cells should be carried for a maximum of 25 passages from the original stock. Always do experiments in a 20 passage window. Day1: 1. Inspect the culture for contamination. This is most easily visible because of pH changes in the phenol red of the media. Cells should be 70-80% confluent. 2. Prewarm Hela media and 0.05% Trypsin-EDTA in the 35C water bath for 15-20 minutes. 3. Aspirate the old medium from the flask with a sterile pipet (I use a 1ml tip on the vacuum) 4. Wash the cells with 5ml of calcium-magnesium free phosphate buffered saline (CMF-PBS) to wash away residual medium. 5. Add 3-5ml 0.05% trypsin-EDTA (TE) and evenly disperse over the surface by rocking. After thawing, the trypsin can be stored at 4C for up to 2 weeks. Do not freeze thaw. However I have been keeping my trypsin stocks at 4C for months with no detectable problems. 6. Place the flask into the incubator with the cap screwed on tightly. Remove the cells every 2 minutes to observe the detachment 7. When cells are detached, add 5ml of new media and rinse the surface of the flask by rocking. 8. Pipette up and down to dissociate cell clumps. Check briefly under the microscope to confirm that cells are dispersed in a single-cell suspension (> 80% of the cells) and that any remaining cell clumps have 5 cells or less. If not, pipet up and down 3 more times at most. Minimize foaming since it is very hard on the cells. a. I actually haven’t ever done the above step 8. I always take a 10ml pipette and mix the cells by pipetting up and down 5 times. I then take this 10ml volume of cells and centrifuge it in a 15ml graded conical tube for 2 minutes at <= 100g. I use the size of the pellet from this to decide how many cells to transfer into the next T75 flask. 21 b. I remove the media/trypsin from the pellet with the vacuum and resuspend the cells in 10ml DMEM+FBS+NEAA (Dulbecco’s Modified Eagle Medium, Fetal Bovine Serum, Non-essential amino acids). c. If the cells are going to be used for transfections/siRNA where the number of starting cells is especially important, I will then remove 5ul of this suspension and place it on the hemacytometer, view the cells under the microscope and calculate the cell density. This is done by counting the number of cells inside the marked box and multiplying by 10 to get cells / ul. (The hemacytometer has documentation showing how, as does my notebook). For most ‘normal’ cell cultures this appears to work out to be approximately 150ul / well of a 6 well plate in order to get 5x10^4 cells. d. Aliquot 10-12ml of DMEM/FBS/NEAA to a prelabeled T75 flask, 1ml/well of a 24 well plate, 2ml/well of a 6 well plate respectively depending on requirements. e. Add an appropriate volume of cell suspension to get 8x10^4 cells in the T75 flask (75cm^2) (For the T75 I don’t think this needs to be super exact, so I will usually had 3 drops.) 9. First mix the cells with the flask upright. Then lay it down and swirl it in a figure 8 pattern. It is very important to get the cells evenly distributed. (The reason for this is because if they are not evenly distributed, they will not be able to communicate properly with each other and will thus grow poorly). 10. Add 4ul of G418 drug / 1ml of medium for the T75 flask. If using these cells for transfections, do not add drugs. 11. Return the cells to the incubator. Day 3. Cells are fed with DMEM/FBS. 1. Aspirate medium with a sterile tip and add 10-13ml new medium and drug. Day 5 Treat cells as on Day 1, but seed at 4x10^4 cells / cm^2. If the cells are > 100% confluent, adjust the seeding density. Thawing HeLa cells. It is important to have everything ready before retrieving the vial from the LN2 to minimize the time the thawed cells are exposed to high concentrations of DMSO. 1. Dispense 10ml of media into a sterile 15ml tube and warm in the 37C waterbath. 2. Add 10-12ml of fresh media to a pre-labeled T75 flask. 3. Remove the frozen vial from LN2 and thaw it in the 37C waterbath by gently agitating. Keep the cap out of the water to avoid contamination. 4. Spritz the vial with 70% ethanol. 5. Remove the cell/DMSO suspension from the vial with a 1ml pipet and add it to the 10ml warm media. 6. Pellet the cells by centrifugation 2m @ 100g. 7. Aspirate the medium 8. Resuspend the cell pellet in 1ml fresh DMEM/FBS gently. 22 9. Add the cell suspension to the T75, swirl in a figure 8 pattern. 10. The following day, remove the old medium and replace with fresh medium. Passage cells when they reach 80-90% confluency. Freezing down HeLa cells. 1. I have been following Sergey’s protocol with a couple of exceptions: a) we use t75 flasks and so cannot get 5-6 vials. In addition, I have been using all of the remaining cells from my cell splitting for freezer stocks. 2. With that in mind, I perform the cell splitting as above and perform this task with the remaining cells in solution of fresh media. 3. Remove the HeLa freezing medium from the cold room. (3ml DMSO to 37ml DMEM/FBS) 4. Remove a cryogenic tube from the stock and label with passage number, date. 5. Spin the cells remaining from the splitting down 2 minutes @ 100g. 6. Remove the medium and add 1.5-2.0ml freezing medium to the pellet and move it to the labeled cryogenic vial. 7. Place the cryogenic vial in a Styrofoam box with a lid in the -80C freezer overnight. Transfer the vial to LN2 storage the following day. Media Preparation: DMEM+10% FBS FBS is stored at -20C and needs to be aliquoted after thawing. To thaw: Remove the serum from the freezer and place it overnight at 4C. Transfer it to a 37C waterbath and agitate periodically. Do not keep FBS at 37C longer than necessary. Set up 50ml sterile tubes in the hood, label caps with FBS and expiry date. Aliquot 27ml FBS / tube and seal with parafilm. Store the aliquots at -20C. New media stock procedure: 1. I now like to aliquot 50ml bottles of DMEM+FBS+NEAA and then add Penn/Strep as needed, 20ul for every 1ml of media. 2. Thus for each new bottle of 500ml DMEM I add: a. 50 ml (I usually add a little extra, 55 ml) of FBS, also pre-aliquotted in 50 ml bottles. b. 5 ml (I usually add 6 ml in reality) of 100x NEAA c. 10 ml (I usually add 12 ml) of 200mM glutamine. I also keep a series of bottles of DMEM with no additions for transfections. These are labeled “DMEM.” Then label the 50ml conical tubes “DMEM +” and place in a plastic bag in the 4C refridgerator. If a bottle is named “DMEM++”, it also contains Penn/Strep to 1x. 23 Trey’s current Transfection protocols When performing transfections it is now important to first consider the experiment. These fall into a few general categories, and require a bit of explanation. siRNA: synthetic precursors of silencing RNAs. These come as lyophilized double stranded molecules with a 1-2 bp overhang on each side. I like to suspend them to [20nM], which makes aliquotting experiments easier. Thus far we have purchased these entirely from Qiagen. Interestingly, if you want the actual sequence for any siRNA, you must call them and prove that we bought it. miRNA: Synthetic microRNAs, these are single-stranded stem-loops which are ready to be processed by Argonaute and friends. I like to suspend these to [5nM], thus far we have gotten them from Millipore and Qiagen. Plasmids: Well known, the only caveat is the promoter. If you are performing steady-state qPCR of Dr. Brewer’s rabbit β-globin, then the cells into which to transfect plasmids should have an actively transcribed transactivator protein (See the mammalian qPCR section for more information). If you are doing dual luciferase, it is usually safe to assume that an endo-toxin free miniprep contains 200ng / ul. For all other uses it is important to quantify the DNA, I prefer the nanodrop for this. 1. Plasmid transfections alone (Dual luciferase) 2. Double plasmid transfections (qPCR with plasmid reference) 3. siRNA alone 4. siRNA + plasmid(s) 5. miRNA alone 6. miRNA + plasmids Plasmid transfection alone is performed by following the Fugene6 protocol to the letter. (Fugene 6 is now sold by Promega). Briefly: Trypsinize and split cells into 6-24 well plates. Let grow to 50-75% confluency. If using 24 well plates, suspend in 500-1000ul DMEM+ (I use 500). In eppendorf tubes in the hood mix 20ul DMEM(no additives!), 0.6ul Fugene, and 300ng plasmid for every 24 well. Let sit 30-90 minutes. Add dropwise to each well of the plate. Wait 1-2 days. Assay. For siRNA or miRNA alone, I prefer the ‘HiPerFect’ reagent. It seems to be the least toxic. However Lipofectamine is also awesome. I again follow the literature protocol pretty explicitly. Briefly: Trypsinize and split cells into 6-24 well plates. Assuming 24 well plates, I add 40,000 cells to every well (counted by hemocytometer) and immediately RNA transfect using the amounts of RNA specific for the reagent chosen. Wait, assay. Lipofectamine is the easiest to aliquot: 2 tubes for every RNA species. 50ul of media in each tube / well of plate transfected. Label 1 tube ‘M+Lipo’ and 1 ‘M+RNA’. Add 1ul Lipofectamine / plate well to the first, and 20pMoles RNA to the second (1ul of [20uM] stock) / plate well. Let sit for 10-20 minutes. Mix together. Let sit 30-90 minutes. Drop on cells, wait, assay. For troublesome plasmids/RNA/mixtures. The available parameters to play with are: Time before transfection, time with transfection reagent, amount transfection reagent, amount of [R|D]NA. 25 Dual luciferase with the Simon Lab Luminometer First a quick table of the dual luciferase plasmids in our lab: Plasmid # pJD375 pJD376 pJD377 pJD378 pJD431 pJD432 pJD433 pJD642 pJD643 pJD644 pJD387 pJD651 pJD650 pJD653 pJD652 pJD449 pJD419 pJD420 pJD421 Auxotrophic Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Ura3 Leu2 Leu2 Leu2 Tests Readthrough -1 PRF +1 PRF -1 PRF nonsense nonsense nonsense missense missense missense PTC control R218C R218S R218T R218T -1 Readthrough -1 +1 pJD633 pJD634 pJD635 pJD636 pJD677 pJD676 Trp Trp Trp Trp Trp Trp Readthrough -1 +1 -1 missense missense Insert LA Ty1 HIV UAA UAG UGA UCU AGC AGU SARS LA Ty1 LA Ty1 HIV Near Near g The first question to ask: working with yeast or mammalian cells? 26 If yeast then DO NOT use the detergent based passive lysis buffer. It foams up and leads to significant variability in reads. We used a PBS based lysis buffer along with bead beating to pop our yeast for dual luciferase. This contains the following in 30ml: 3ml 10x PBS, 0.2ml optional protease inhibitor cocktail, 0.2ml 150mM PMSF, water; made fresh just before lysis. The 150mM PMSF is made by adding 0.2613g of PMSF into 10ml Methanol. PMSF is extremely hydroscopic, find it in the dessicator. 10x PBS in turn is made with: 30.47g Na2HPO4, 0.155g NaH2PO4, 2.19g NaCl, pH this to 7.4 with HCl, add water to 250ml. Autoclave it. The protease inhibitor contains 1ml of: 0.15ml Aprotinin (2mg/ml), 0.15ml leuprotinin (1mg/ml), 0.15ml Pepstatin A (1mg/ml), 0.55 ml water. Yeast Lysis 1. Grow overnight cultures to exponential growth (This varies from an OD of 0.7-2.0 depending on strain. Different strains require different ODs, do a growth curve. 2. Harvest cells by spinning down gently in the Sorvall (1200 rpm for 2 min) and removing media. 3. Wash the cells with 1ml lysis buffer and move them to a fresh eppendorf. 4. Spin them gently and remove lysis buffer. 5. Add 500ul fresh lysis buffer and 500ul glass beads and chill on ice/cold room for 5 min. 6. Vortex in the cold room at minimum 5 minutes or bead beat them 5 minutes. 7. While they are lysing, Start melting an aliquot of the Luciferase Assay Buffer. When it is ready, Make the LARII solution by “Resuspend lyophilized ‘Luciferase Assay Substrate’ to ‘Luciferase Assay Buffer’” This solution may be stored in the -80 for up to a year. 1. For every 20 wells to assay on a plate, 1ml of Stop and Glo is required. Add 20ul of the Stop and Glo substrate (the flammable yellow goop) to 1ml of the Stop and Glo buffer. (I prefer to pre-aliquot 1ml eppendorf tubes). 2. Clarify the lysates by spinning 5min in the cold room. 3. For each well of the plate, use 20-100ul (I like 50ul) of lysate. The method of laying out the plate matters, that will be explained after lysing mammalian lysates. If working with mammalian cells, the process is simpler: Mammalian Lysis 1. 2. 3. 4. Make a 1x lysis buffer from the 5x supplied by Promega. Remove the medium from the cells. Add 200-500ul lysis buffer / well. Rock the plate at RT for 5 minutes. Plate Layout Calculate how much LAR and Stop/Glo will be required before laying out your plate! 27 The plate to use for these experiments is a white, flat bottomed, 96 well plate. Currently they reside in 2136 on the top shelf above the 30 well centrifuge. I usually use 50ul lysate and 50ul of each reagent / well of the plate. The plate reader reads starting at A1 and goes horizontally until the end of the defined read, then B1 etc… In addition the plate reader has functionality to pre-fill the lines with reagent, unfortunately this process wastes about 1ml of each reagent (that is 20 reads!) To get around this problem, I do not use that functionality but instead leave the A row blank, but tell the machine to read it. Starting at A10 reagent will start to get into the wells. I usually start my lysates at B1. I prefer to do 1 replicate of every lysate B1-Bn, then start a second replicate immediately thereafter, and repeat until the plate or lysates are exhausted. I assume the number of desired wells is (# lysates * 4 replicates) and add 2-5 wells. Given this number I aliquot the amount of LARII and Stop/Glo in separate tubes (usually 15ml conical). Luminometer Usage 1. Check in the day before if you can. 2. Boot the machine, choose a protocol, luminescence, dual luciferase 2 injectors. There are 3-5 places on the screen to check out a. The amounts of each reagent to aliquot are default 100 μl, change them(likely to 50 μl) b. The machine by default reads every well of the plate, change that if need be. c. Activate the PMT d. Go to Setup and perform a flush cycle of the machine before performing an assay. This consists of letting it squirt water, ethanol, water, and air into the waste tray. 3. Start the read. It will take about 30 minutes for a full plate. It will ask you for a name of the file before it starts up, choose something unique. 4. When finished, use the USB hard drive to collect your data 5. The data will be in a comma separated format. a. Perform another water,ethanol,water,air flush of both injectors as per step 2d above. 6. The calculations for % Frameshifting are generally pretty easy. a. You are aiming for Experimental:(Read1/Read2) / Control:(Read1/Read2) b. So calculate the average ratio of (Read1/Read2) for each lysate type. Identify your control beforehand, and divide each experimental by the control for % PRF. c. Calculate the standard deviation for those reads. d. Calculating error bars are a bit more difficult: For the standard deviation of the ratiometric mean as an error bar: Experimental Var == (standard deviation of experimental ratio squared / the ratio value) squared Aliquotting Promega materials Promega ships 10 bottles of the following: 1. Freeze dried Luciferase assay substrate (brown opaque bottle) 28 2. Frozen liquid smelly Luciferase assay buffer (super light yellow or clear) 3. Frozen Stop + Glo buffer (Clear) 4. Stop and Glo substrate in small eppendorf tubes (bright yellow and flammable) If we are using the default dual luciferase protocol for the Simon lab luminometer, then 1ml of each mixed reagent is sufficient for 20 wells of a 96 well plate. The mixed LARII (Luciferase assay buffer + substrate) is stable for 1 year at -80C, but is freeze/thaw sensitive. The mixed Stop+Glo is usable for 1 day. The Stop+Glo buffer is stable at -20C for 1 year. Aliquotting strategy: Make 1ml aliquots of LARII to store in the -80. I (Trey) keep them in the far left Styrofoam box on top of the 3rd shelf in the -80. To do so, I pre-label 11-12 eppendorfs with ‘LARII’, I should add the date, but haven’t thus far, I think I will from now on if other people will use this protocol. I then thaw the Luciferase assay buffer on my wrist and add it all to the brown luciferase assay substrate bottle and mix. I then aliquot it to the eppendorf tubes and place it in the -80 box. At the same time, I take 1 bottle of Stop+Glo buffer, and do the same thing with 10-11 eppendorf tubes labeled ‘S+G.’ Each tube therefore should be ready to have 20ul of Stop and Glo substrate added for every 20 wells of a 96 well plate. 29 Flow Cytometry and Fluorescence Activated Cell Sorting Short version: To assess coreceptor levels, TZM-bl cells were harvested with (Invitrogen #13151014) a PBS dissociation buffer without enzyme. They were washed and stained with: primary antibody Mab hCCR5 (aidsreagent part numbers: 45531.111, 45523.111), which contains the IgG2a isotype from a Balb/c mouse; and secondary antibody Goat Anti-Mouse IgG2a (γ2a chain specific) conjugated to Fluorescein (Southern Biotech #1080-02). Fluorescence intensity was analyzed by flow cytometry of 10,000 events (using a BD FACS Canto II). Long version: This process is intended to be very similar to that used for Western blotting. Transfections are performed to repeat conditions used for mRNA abundance, thus [10 nM] siRNA over two days. Some caveats: Most printed protocols are for intracellular proteins. As a result they permeabilize the cells with Tween-20, then fix them with methanol or formaldehyde. In this particular case, my protein of interest is on the outside of the cells and so I should maintain them with only PBS/FBS. 1. Wash cells in PBS based dissociation buffer. Determine the number of cells with the hemacytometer. Resuspend cells to 1-5x106 cells/ml in ice cold PBS, 10% FBS, 1% NaN3. a. It appears that this particular primary antibody binds much better after permeabilization. This may be performed with PBS-Tween. 2. Aliquot cells to 100 µl for each experimental tube. 3. Add 0.1-10 µg/ml primary antibody. (Common dilutions of antibody are between 1:200 – 1:10,000) 4. Incubate cells at 4° 30-60 minutes in dark. 5. Wash cells 3 times by gentle centrifugation (400 g for 5 minutes) and resuspend in ice cold PBS, 3% FBS, 1% NaN3. 6. Dilute the fluorochrome-labeled secondary antibody in PBS, 3% FBS at the optimal dilution (likely 1:500) and resuspend the cells in this solution. 7. Incubate cells at 4° 20-30 minutes in dark. 30 8. Wash cells as per step 5. 3 times by gentle centrifugation (400 g for 5 minutes) and resuspend in ice cold PBS, 3% FBS, 1% NaN3. 9. Store cell suspension immediately at 4° in dark. Analysis should ideally be performed immediately. If necessary, the cells may be fixed for short-term storage. 1. Add 1 ml ice cold acetone (methanol is allowed as well) to each sample. Mix gently. Place at -20° for 5-10 minutes, centrifuge 400 g for 5 minutes, and wash twice in PBS 1% FBS. The preceding protocol comes from: http://www.abcam.com/index.html?pageconfig=resource&rid=11381 It has an interesting omission which is performed by the Mosser lab: Blocking with PBS with 5% FBS and the FC Block reagent. (FC block is an antibody against the crystallizable fraction of antibodies – this consists of the homogenous “stem” of antibodies) If performing this step, a 1:100 dilution of FC block antibody is applied between steps 2 and 3 and incubated at 4° 30-60 minutes. FACS Canto instructions: Caveats: 1. Do not change parameters/gates while the machine is recording a sample. This may lead to a system lockup. 2. The loading system (aspirator arm) is sprint loaded and will shut if one is not careful. 3. Between reads, allow the aspirator arm to perform its cleaning operation. a. When it runs, it sets the arm down on its little pad, expels some liquid, and sucks it backup. Thus if you don’t let it complete, it will expel into your tube. 4. Start each read with 5-10 seconds of acquisition before recording. This allows the sample flow to stabilize. 5. Do not force the aspirator arm. Allow it to finish each operation before changing its state. 6. Do not allow any air bubbles to form. This would (for example) happen if the tube is left unattended. Keep an eye on the level of sample in the tube. Stop reading 31 before it reaches the convex portion. Alternately, set a stop-time in the system panel. Usage: 1. Log in as dinman 2. Copy and paste the HeLa experiment under ‘Folder_001’ to a new experiment. 3. Currently a series of tubes will automatically load including: a. 2ab: Baseline control of only secondary antibody on cells which have not been transfected b. 2ab 1:50 – 2ab 1:50,000: 1:10 dilution series of primary antibody and secondary antibody. Performed using control cells (scrambled or untransfected usually), secondary set to 1:500 c. Cells only: No antibody at all. This, along with the PI stain provides a measurement of how well the cells fared in collection. d. Scrambled: A scrambled siRNA control, usually with 1:5,000 primary antibody, secondary 1:500 e. Experimentals: 1:5,000 primary and 1:500 secondary. 4. Take the 500 µl of a given sample and load it into the 12x75 mm Fisher tube (14956-3c) 5. Add a small amount of PI dye. Allow it to stain for ~ 1 minute. This may be performed for future runs while the current run is going. 6. Load the tube and hit ‘Start acquisition’, wait a moment then, ‘Start Recording.’ 7. Allow the run to continue for 240 seconds or 10,000 events, whichever comes first. 8. Stop acquisition, remove tube. Wait for cleaning process to complete. 9. Repeat to step 4 until all samples are used. Data usage: 1. All operations are manipulated via the ‘Global Worksheet.’ Changes to this sheet are propagated throughout the experiment. It currently contains 6 views of the cells: 32 a. FSC-H vs FSC-A provides a view of cell size b. SSC-A vs FSC-A provides another view of cell size and has the first logical gate which separates small (presumably dead) cells from the larger (presumably viable). c. FSC-A vs FITC-A provides the first view of size vs antibody d. Count vs FITC-A provides a histogram of fluorescence intensity -- I just added a third logical gate to this, which still needs to be correctly set, but should eventually provide my difference vis a vis antibody abundance. e. Count vs. PI longred provides a histogram of viability -- this currently has the second logical gate, providing a means to separate cells which exclude PI f. PI longred vs FSC provides a view of viability on both axes. 2. Cytometer voltage calibration. Currently 4 wavelengths are being monitored: a. FSC multiplier: 0.3 b. FSC Voltage: 212, not log c. SSC Voltage: 203, log d. FITC Voltage: 362, log e. Propidium Iodide Voltage: 275, log 3. The key (still not understood by me) is to adjust these parameters to maximize the separation of viable samples to non-viable (for FSC/SSC/PI) then for positive hits vs negative hits (for FITC). In order to do so, start acquiring, then change the voltage +- 50 and watch the various histograms. If they get closer, redo. 4. Shutdown: 10% bleach in sample tube. "Cytometer -> Cleaning mode -> Clean flow cell" x 4. Remove Tube. Fluidics shutdown. hit ok. Do not log out, or shutdown will halt. 33 Alternate FACS cell staining method (Song lab) Materials PB for Confocal (also FACS): 600 ml DMEM 60 ml FBS (10%), filtered 6 ml 1 M HEPES (pH 7.6) 6 ml 1 M glycine 3 ml 10% saponin Aliquot to 50 ml conical tubes and freeze down at -10° PB specifically for FACS (no DMEM): 1 ml FBS 1 ml 1M HEPES 1 ml 1 M glycine 500 µl 10% saponin Bring up to 100 ml with 1x PBS Optionally add 0.05% sodium azide. Protocol 1. Dissociate cells with trypsin free dissocation buffer (PBS + EDTA) 2. Spin cells down 1,100 g 5 minutes. 3. Fix cells with 4% paraformaldehyde (PFA) for 20 minutes at RT. 4. Wash cells with 1x PBS 3 times and with spin down at 1,800 g 5 minutes. 5. Permeabilize cells with PB for 15 minutes 6. Incubate cells with 1 µl primary antibody (undiluted) for 30 minutes at RT in 50 µl PB 7. Wash cells 3 times in PB with 5 minute 1,800 g spin 8. Incubate cells with 1 µl secondary antibody (undiluted) for 30 minutes at RT in 50 µl PB 9. Wash cells 3 times in PB with 5 minute 1,800 g spin. 10. Wash cells 3 times in 1x PBS with 5 minute 1,800 g spin. 11. Fix cells with 4% PFA for 20 minutes at RT 12. Wash cells 3 times in 1x PBS with 5 minute 1,800 g spin. 13. Perform FACS analysis immediately. 34 35 Western Blot Short version Cell lysates were prepared from TZM-BL cells transiently transfected with siRNA against hSmg1, hRent1, hUPF2 and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting as previously described (Vila-Coro et al., 2000) with changes as per (Ribardo et al., 2004). Westerns were performed with antibodies against hCCR5(BioLegend Mouse IgG anti hCCR5 #321702) followed by secondary antibodies conjugated to horseradish peroxidase (HRP) (SantaCruz Goat IgG-HRP #sc-2005). The lysates were also probed with anti- tubulin antibodies (Mouse IgG anti Tubulin, a kind gift from the Song laboratory) followed by secondary antibodies against mouse IgG conjugated to HRP. (unsuccessfully with SantaCruz #sc-20227 Lot # A0307 Goat polyclonal IgG followed by SantaCruz #sc-2020 Donkey anti-goat IgGHRP) After probing, blots were exposed using Luminol (SantaCruz #sc-2048) and a Fujifilm CCD camera. Long version http://biologicalworld.com/westernblot.htm RECIPES: when working with proteins, try to use nanopure water 10% SDS 10g SDS up to 100mL water Buffers for Making Gels Lower Buffer (for the separating gel) 1.5M Tris 36.4g pH to 8.8 with 6M HCl until nearing desired pH 0.4% SDS 8mL of 10% SDS up to 200mL with water Stacking (Upper) buffer (for the stacking gel) requires fair bit of HCl so start with ~70mL water when add Tris 0.5M Tris 6.06g pH to 6.8 with 6M HCl until nearing desired pH 0.4% SDS 0.4g up to 100mL with water 30% Acrylamide* Acrylamide 30g Bis 0.8g up to 100mL with water wrap aluminium foil around bottle because it is light sensitive 36 Making SDS Polyacrylamide Gels* (once the last ingredient, ammonium persulfate, is added, the gel will begin to polymerise) Separating (lower) Gel - 10% acrylamide (change the proportions of water and acrylamide if different from 10% acrylamide) Lower buffer 1.9mL Water 3.1mL Acrylamide 2.5mL TEMED 10uL (add TEMED in the fume hood) Ammonium Persulfate 20uL Total 7.5mL *Note that acrylamide in its unpolymerized form is a potent neurotoxin and that gloves must be worn for making gels, setting up the tank for running the gel, and during transferring of the gel Stacking Gel (for all percentages of lower gel, use this upper gel) Upper Buffer Water Acrylamide TEMED Ammonium Persulfate Total 1.25 mL 3.25 mL 0.5 mL 10uL (add TEMED in the fume hood) 20uL 5 mL 10x Electrophoresis Running Buffer (for the tank) Tris 121.4g Glycine 567g SDS 40g Water up to 4L pH 8.3 without adjustment 4x SDS Loading buffer Glycerol 4mL (put an empty flask onto balance and weight out glycerol by pipetting) SDS 0.4g Upper buffer 5mL up to 10mL with water · heat while stirring to dissolve the SDS (don’t make the solution boil when heating), and add enough bromophenol blue to make the 1x solution dark enough for easy monitoring when running on a gel · store at room temperature 1x Coomasie Blue Dye (for staining gels that are not used for transferring, or for ensuring that no protein is left after transfer) Isopropylalcohol 25% 500mL Acetic Acid 10% 200mL R250 Coomasie Blue 0.025% 0.5g 37 Water 1.3L (destain is 10% Acetic Acid) · stain 4 hours to ensure all the protein is stained · destain long enough so that the background is clear (a piece of foam or paper towel may be added to the solution to speed up the destaining) 10x Towbins Transfer Buffer (for semi-dry transfer of protein from gel to blot) 250mM Tris 15.1g 1.92M Glycine 72.0g Water up to 500mL pH 8.3 without adjustment 10x Phosphate Buffered Saline—PBS (for removing methanol from blots) NaCl 1.37 M for 10x KCl 2g 27mM for 10x Na2HPO4 80mM for 10x KH2PO4 20mM for 10x water to 1L pH of the 1x PBS should be 7.4 PBS Tween—PBST (for washing blots) PBS 2L Tween-20 (Brown bottle) +1mL by pipetting it in with pipettor or plastic bulb stir half an hour Amido Black Stain (for viewing all proteins non-specifically after blotting) Methanol 40mL Acetic Acid 10mL Amido Black 10B 0.1g Up to 100mL Western Blot protocol Making the gel 1. Use alcohol and Kimwipes to wipe the glass, and then set up the rest of the apparatus a. For small proteins, use a higher percentage gel 2. Add all ingredients except ammonium persulfate; before adding ammonium persulfate, if the weather is cold in the room, you may wash the solution with hot/warm tap water to help the polymerization 3. Invert tube gently after adding all components 4. Add it up to the correct mark 5. Add t-butanol 6. Remove t-butanol, wash with water, and wipe with filter paper; put in comb 38 7. Make upper gel and gently shake 2x 8. Add upper gel 9. Remember to keep tubes to check if they’ve polymerised (cap the tube after pouring and see if the material in the tube has polymerised) 10. Label the lanes of the upper gel with a marker if required Preparing and Loading Samples 1. Set up the gel in the tank and make sure the buffer isn’t leaking (if it is, it means that the glass plates are not slightly protruding up from the screws on both ends) 2. Pour in some running buffer to prevent drying of the gel 3. For each sample, use 1/3 the volume of 4x loading dye (e.g. if have 30uL of sample, add 10uL of 4x loading dye) 4. If running samples in reduced form, add 5% beta-mercaptoethanol in the fumehood 5. once the loading dye has been added, do no place on ice, or the SDS will precipitate out Running and transferring the gel 1. Check no leaking and it’s running 2. Set for anywhere from 20-30mA, and all the voltage it needs 3. Keep an eye on it to ensure there is current 4. Cut out filter paper and nylon—eight sheets of filter paper for each blot 5. Mark the front of the nylon (make a cut) FOR NITROCELLULOSE BLOTS DON’T WET WITH METHANOL, OR ELSE IT’LL DISTINTEGRATE IN YOUR HANDS 6. Wet the nylon with methanol 7. Immerse gel, filter paper, and nitrocellulose in transfer solution (20% methanol + Towbins 1x); use a lower percentage of methanol for larger proteins (e.g. 7%) 8. Proteins in SDS are negatively-charged, so place sandwich as follows (flatten with each layer except for gel layer because gel is fragile) Black plastic Pad Whatman Gel PVDF Whatman Pad White plastic a. 3 pieces filter paper b. nylon c. gel (proteins in SDS are negative) d. 3 pieces filter paper 9. For transfer conditions, check with manufacturer. We normally set at 20V, 0.300A, 20 minutes (time varies—larger proteins require longer time) 39 10. Rinse blot with distilled water and hang to dry 11. Wipe the transfer apparatus with plenty of water 12. Dry the transfer apparatus more quickly, wipe with some towels, and then fan the remaining liquid with a piece of paper towel Blotting 1. Wet a dried blot with methanol if the blot is made with PVDF, or soak in PBS if blot is made from nitrocellulose (note that methanol is toxic, so wear gloves and don’t inhale it) 2. Rinse ~4x with PBS to get rid of the methanol 3. Place the blot face up for blotting 4. 1% skim milk (made in PBS) 10 min. (some proteins require a higher percentage of skim milk e.g. 5% and incubation in the cold room for longer e.g. 30min.) 5. 10mL total--Primary antibody diluted to the appropriate concentration in PBS (+0.2% milk) 1hr 6. 3x 10min. each PBST 7. 1:5000 dilution of secondary (choose the right secondary—either goat anti-rabbit, or goat-anti mouse), 30 minutes 8. Wash ~5x PBST 5-10 minutes each Viewing 1. 2. 3. 4. 5. mix equal amounts of solution A & B of ECL (~1.5mL each of A & B) rock manually for 1 min. wrap Saran Wrap around blots take off gloves when handling film turn off all lights (can leave a dim red light on) when removing film from the box, and place remaining film back into box before dealing with the newly-removed piece of film 6. place film on top of blot in the film cassette, making a note of the orientation of the film 7. expose for 15sec -2 minutes, and if required, use another film and expose for anther 20 min. 8. develop Nonspecifically staining the blot 1. rinse blot with distilled water several times to remove the ECL 2. invert amido black bottle a few times, and pour some into a petri dish with the blot in it 3. rock blot a few times 4. pour amido black back into bottle 5. rinse with distilled water several times to destain the blot 6. hang blot to dry and place in notebook If blot turns dark blue irreversibly, dry or put in PBST and then 100% methanol 40 Bacterial Transformation Short version: Escherichia coli strain DH5α was used to amplify plasmid DNA. Transformations of E. coli were performed as described previously using the calcium chloride method(Sambrook & Russell, 2001). Long version: 1. Remove DH5α cells from the -80° freezer and defrost on ice for 2-5 minutes. 2. Aliquot 20-50 µl DH5α for each transformation. 3. Add 0.1-2 µl plasmid, incubate on ice 10-100 minutes. 4. Plate on LB plates with appropriate selection (usually Carbenicillin). Low efficiency transformations may require additionally incubating the bacteria at 37° for 1 hour with 200-800 µl LB media. In addition it may be necessary to add 1 µl βmercaptoethanol to the cells before incubating with plasmid. For very low efficiency transformations, consider using the XL cells from the stratagene site directed mutagenesis kit. 41 Yeast Transformation Short version: Yeast cells were transformed using the alkali cation method(Ito, Fukuda, Murata, & Kimura, 1983). Long version: 0. Double check yeast genetic background and plasmid to ensure everything will work. 1. Grow 2-3 ml yeast overnight in appropriate non-selective (for your plasmid) media. 2. Start salmon sperm ssDNA boiling early. 3. Aliquot yeast into 1.6 ml eppendorf tubes and spin down at < 2,000 rpm. If using the sorval, I never go higher than 1,200 rpm. 4. Remove media and resuspend in 200 µl 0.1 M LiOAc/TE 5. Spin again, remove LiOAc/TE, resuspend in ~ 100 µl 0.1 M LiOAc/TE 6. Add 15 µl salmon sperm ssDNA / transformation. 7. Add 5-20 µl plasmid / transformation 8. Add 500-700 µl PEG/LiOAc/TE 9. Incubate 90-900 m at 30° (Longer is generally better). 10. Heat shock at 42° for 15 m. 11. Pellet cells, remove PEG/LiOAc/TE, add 100-400 µl water. 12. Plate and let grow 1-4 days. Caveats: All amounts are negotiable. But be aware that LiOAc is mildly poisonous. I never take them for more than 20 minutes at 42°. If the cells are sick then I sometimes do the transformations entirely at RT. For 10 ml LiOAc/TE/PEG: 8ml 50%PEG 3500MW + 1 ml 10X TE + 1 ml 1M LiOAc. 42 Cell culture Short version: HeLa(Gey, Coffman, & Kubicek, 1952) (ATCC), HeLa Tet-Off(Gossen & Bujard, 1992) (Clonetech), HeLa TZM-BL(Platt, Wehrly, & Kuhmann, 1998) (aidsresearch.org, and kindly provided by Dr. J. DeStefano), CHO(Puck, Cieciura, & Robinson, 1958) and Vero(Sheets, 2000) cells were cultured according to manufacturer’s instruction. Long Version: HeLa derived cells and Vero cells are maintained at 37° in 5% CO2 using DMEM supplemented with 10% FBS, 1x non-essential amino acids, and [4 mM] glutamine. 1% Penicillin/Streptomycin may be used when not performing sensitive assays. CHO cells are maintained in the same fashion except using supplemented MEMα. HeLa Tet-Off cells were cultured with 200 µg/ml G418 in order to maintain the transactivator protein. Passage: Cells are best passaged when growing exponentially, 70-80% confluency. HeLa derived cells should not be maintained for more than 25 passages, and experiments should be performed before 20 passages. 1. Check for contamination. This is generally made easy by phenol red in the media. 2. Pre-warm fresh media and 0.05% Trypsin-EDTA at 37°. 3. Aspirate the old media from the flask with a sterile pipet. Wash cells with 5 ml PBS to remove residual media. 4. Add 3-5 ml 0.05% Trypsin-EDTA and evenly disperse by rocking. (Storage note: After thawing, trypsin may be stored at 4° for up to 2 weeks, do not freeze thaw. In practice, trypsin may be stored significantly longer) Incubate at 37° 5-10 minutes to detach cells. 5. Add 5 ml fresh media to wash cells from the plate surface, pipette to dissociate cell clumps. (Avoid making foam, it kills) Remove to 15 ml conical tube and spin down 2 minutes at < 100g. 6. Remove media and resuspend in 1 ml DMEM. Add 1 µl cells to hemacytometer with a small amount of trypan blue to count viable cells. (100 undiluted cells in the square on our hemacytometer corresponds to 1,000 cells / µl in the conical tube.) 43 7. Aliquot cells according to need. (30,000-40,000 cells / well of 24 well plate, 100,000 cells on a T75 flask) 44 Plasmid Construction Short Version: Synthetic oligonucleotides (IDT DNA) used in this study are listed in () and plasmids are listed in (). Insertions were amplified using PCR and ligated into appropriate backbone plasmids. Mutagenesis was performed using QuikChange (Agilent) site-directed mutagenesis kits. Clones were confirmed by sequencing (Genewiz). Long Version: Plasmid construction: 1. The PRF signal from Homo sapiens CCR5 was amplified from pCMV-XL4 (pJD819) containing the CCR5 open reading frame (Origene) using oligonucleotides with Bam HI and Sal I restriction sites. PCR products were ligated into p2luci (pJD175e)(Grentzmann, INGRAM, KELLY, GESTALAND, & ATKINS, 1998) 2. Oligonucleotides encoding the -1 PRF signals from the hIL2, human and mouse IL7, hIL8, hIL8, hIL22, and hIL27 receptor chains were purchased containing 15-18 bases of internal overlap, PCR extended into a single Bam HI and Sal I containing product, and ligated into p2luci. 3. pTRE-Rβ (pJD976), pTRE-Rβ-ARE (pJD975), and pTET-Off (pJD979) were kind gifts from Dr. G. Brewer(Ysla, Wilson, Brewer, & Riza, 2008). Insertions of the CCR5 -1 PRF signal were performed via a modified version of the “mega-oligo” site directed mutagenesis protocol.(Ke & Madison, 1997). Oligonucleotides (IDT) were chosen to include 23 nucleotides of the β-globin exon 1, 36 nucleotides of Renilla luciferase on the 5’ side; and 23 nucleotides of β-globin exon 1, 15 nucleotides of firefly luciferase on the 3’ side; and amplified from the CCR5 dual luciferase plasmid (pJD827). The resulting 330+ nucleotide oligos were used with pTRE-Rβ template to generate pTRE-Rβ-CCR5 and variants (pJD973, wt CCR5; pJD974, wt CCR5 and ARE; pJD977, synonymous mutation of CCR5 slippery heptamer; and pJD1058, mutation of CCR5 slippery heptamer to GCGCGCG) Oligo-annealing PCR cloning 1. Oligonucleotides are chosen to include an appropriate spacer (often 4A), a restriction enzyme cutting site unique to the cloned sequence and occurring in the multiple cloning site of the backbone plasmid, and 50-75 bases of sequence to be inserted ending in a region which will overlap. 2. In most cases, the 5’ oligonucleotide is chosen to exclude one base immediately after the slippery heptamer in order to put the fragment up to and including the heptamer in the 0 frame, and the following fragment in the +1 frame. The overlapping sequence must be chosen carefully to have a sufficient Tm and properly maintain the reading frame of the upstream and downstream ORFs. Finally, ensure that the 3’ oligonucleotide is the reverse complement of the coding sequence. 45 3. Perform a PCR reaction using only the two oligonucleotides with an extension time short enough to avoid primer oligomerization. The annealing temperature should be 2-4° less than the Tm of the overlapping sequence. 40-60 cycles may be required. 4. The 100-200 base pair fragment should be gel-purified to remove unincorporated oligonucleotides, ethanol precipitated, and eluted in 10-30 µl water. 5. Digest the resulting dsDNA fragment and template plasmid with the appropriate restriction enzymes. 6. Clean the resulting linearized plasmid and fragment before ligation. This may be done by PCR purification, gel purification, or ethanol precipitation. Ideally the final elution should result in a 10:1 insert:vector molar ratio. Using OD260 to quantify the insert:vector at this time is unreliable either because the short fragment has an ill-defined extinction coefficient or because of contaminants from gel purification. Thus it is advisable to quantify by electrophoresis and comparison to a DNA ladder. 7. Ligate the fragment and plasmid and transform for amplification. Because the fragment is so small, I have found that ligation works best when using a T3 DNA ligase overnight at 16°. Mega-oligo SDM 1. Oligo generation is performed via PCR using 10-100 pMoles each primary oligonucleotide and 10-100 ng template plasmid (Generally dual luciferase plasmid). 30-45 cycles of PCR are used with an appropriate annealing temperature (2-6° less than the Tm of the contributing bases, those bases comprising a portion of the dual luciferase plasmid) and a short extension time (30 seconds) at 64-68°. 2. The resulting short double-stranded DNA product is gel-purified and ethanol precipitated and eluted in 5-20 µl water. 3. 50-100% of the purified oligonucleotide is used in the agilent Quikchange SDM protocol without modification. 46 -1 PRF quantification by dual luciferase Short Version: Dual luciferase assays were performed as previously described(Grentzmann et al., 1998) with some modifications. Long Version: HeLa, CHO, or Vero cells were seeded into 24 well plates (3-5 x 104 cells/well) and cultured for 24-36 hours before transfection with dual-luciferase plasmids. Cells were transfected with dual luciferase plasmids using 0.6 µl of the FuGene 6 reagent in 20 µl of DMEM without FBS. 200400 ng of plasmid was used per well and incubated for 20-60 minutes at room temperature. At 24-48 hrs post-transfection, dual luciferase assays were performed using the standard dual luciferase protocol (Promega) with slight modifications using a Turner Biosystems GloMax-Multi Microplate Multimode Reader. Changes to the standard protocol are as follows: lysates were resuspended in 200 μl 1x lysis buffer before reading using 15-35 μl lysate/well, and 50 μl of each reagent/well were used with a 10 second integration and 2 second pause between reads. All assays were performed at least in triplicate, and statistical analyses were performed as previously reported(Jacobs & Dinman, 2004). 47 -1 PRF quantification by in-vitro transcription Short Version: -1 PRF was observed by in-vitro transcription-translation as per (Su, Chang, Chu, Tsai, & Chang, 2005). Long Version: To monitor -1 PRF in vitro, linear DNA templates were prepared by PCR reactions using plasmids pJD175f, pJD187, pJD827, pJD1078, and the T7-Kozak-Renilla/PolyA-Stop-firefly primer set. PCR products were purified by agarose gel electrophoresis. Capped mRNAs were synthesized using the mMESSAGE mMACHINE Kit (Ambion). Transcription reactions (40l) were assembled containing 2µg linearized DNA templates, incubated at 37°C for 4 hours, and mRNAs were purified using a MEGAclear mRNA purification kit (Applied Biosystems). mRNA concentrations were calculated from OD260 readings. In vitro translation reactions were assembled in a total volume of 25µl containing 0.2 to 5µg capped mRNAs using the Retic Lysate IVT Kit (Applied Biosystems). Reactions contained [35S]Methionine (1175Ci/mmol, Perkin Elmer) and the 20x –Met Translation mix provided by the kit. Reactions were incubated at 30°C for 90 minutes. Translation products were resolved through 12% SDS-PAGE, and translation products were visualized and quantified using a phosphorimager. 48 qPCR Short version: qPCR analyses were performed as previously described(Ysla et al., 2008) with modifications. Long version: The dual luciferase readthrough control was used for co-transfections rather than EGFP. RNA samples for qPCR were isolated using the RNAqueous kit (Ambion), digested with rDNAse (Ambion) and analyzed using agarose gel electrophoresis and/or OD260/280 measurements. The remaining samples were reverse transcribed using the iScript cDNA kit (Bio-Rad). The resulting cDNAs were diluted to 1:50-10,000 depending on mRNA concentration. Reactions were performed using 10 μl of LightCycler 480 SYBR Green I Master mix (Roche), 0.2-0.3 μM of each oligonucleotide, 2 μl of cDNA, and water to 20 μl/well. All samples were assayed for genomic DNA contamination by performing the assay using wells containing 1-2 μl of digested mRNA instead of cDNA. Reactions were amplified using either a Roche 480 LightCycler or a Bio-Rad CFX 96 thermocycler as follows: 25⁰C for 10 seconds, 95⁰C for 5 min, followed by 45-60 cycles of 95⁰C for 10 seconds, 52⁰C for 15 seconds, and 72⁰C for 15 seconds. Melting curves were monitored by taking readings every 0.5⁰C from 52-95⁰C. The time-course qPCR analyses were performed with 53⁰C and 54⁰C annealing temperatures and 20 second extension time with no significant changes in results. For qPCR analyses of the full length CCR5 mRNA, assays were performed as described for the β-globin assays, but using oligonucleotides specific for β-micoglobulin and/or GAPDH and CCR5(Mattapallil et al., 2005). Reactions were amplified using the same conditions as for the β-globin constructs except all reactions used 20 seconds at 55⁰C for extension. All assays were performed at least three times. 49 Mammalian time-course assays Short version: mRNA decay time course assays using the tetracycline repressible rabbit -globin reporter were performed as previously described with minor changes(Ysla et al., 2008). Full length CCR5 time course assays were performed as per (Yamazaki & Takeshige, 2008) with modifications. Long version: Time-dependent decay of the β-globin reporter mRNA was assayed by co-transfecting cells with the dual luciferase readthrough reporter (pJD175f) and doxycycline repressible β-globin reporter. After 24-48 hours, fresh medium with 2 µg/ml doxycycline was added. At each proscribed time-point, mRNA as collected with the RNAqueous kit (Ambion). qPCR was performed as per steady-state assays. To monitor time-dependent decay of the CCR5 mRNA, HeLa Tzm-BL cells were first transfected with either scrambled or hSMG1 siRNAs as described above. Forty eight hours after transfection, cells were treated with actinomycin D (10g/ml at time = 0 and and additional 2 g/ml in all following time points) In all experiments, RNA isolations were performed immediately at each timepoint after transcriptional arrest using the RNAqueous kit (Ambion) rather than after freezing samples on dry ice. qPCR was performed as per steady-state assays, using GAPDH as a reference. 50 RNAi Short version: Cells were transfected with oligonucleotides using the HiPerFect transfection reagent (Qiagen), siPort (Applied Biosystems/Ambion), or Lipofectamine-2000 (Invitrogen). Long version: Cells were transfected with siRNA oligonucleotides specific to hUpf1, hUpf2, hSmg1 argonaute, or scrambled oligonucleotides using the HiPerFect or Lipofectamine-2000 (Invitrogen) transfection reagent (Qiagen). Initial transfections were performed at [1, 5,10 nM], and [20 nM] for optimization. The MAPK cell-death positive control was used for optimization as well as qPCR quantification of the targeted mRNA. Most final transfections were performed at [5 nM]. 1. 30,000-40,000 cells were aliquoted (via hemacytometer) in 500 µl of DMEM+FBS in a 24 well plate. 2. For each well, 100 µl of DMEM without FBS and 3 µl of HiPerFect were incubated together at room temperature for 5-10 minutes. siRNA oligonucleotides were added and incubated an additional 15-30 minutes at room temperature. 100 µl of the mixture was added to each well. 3. Media was replaced with fresh DMEM+FBS after 8-12 hours. Assays were performed 36-72 hours after siRNA transfection. When reporter plasmids were employed, they were transfected into cells separately 24-48 hours after siRNA transfection using the Fugene 6 (Roche) reagent or Lipofectamine-2000(Invitrogen). 4. Depending on initial results, a second siRNA transfection is performed 24 hours after the first. hUPF1 is the most likely to require a second transfection in order to have an effect, presumably due to its relatively high abundance(Maquat & Serin, 2001). miRNA transfection Cells were transfected with the following miRNA precursors at [5-30 nM] depending on cell viability: hsa-miR-141, hsa-miR-711, hsa-miR-1224-5p, and hsa-miR-1205 using either siPORT, lipofectamine 2000, or HiPerfect reagent. When performing the miR1224 titration, hsa-miR-1224-5p concentrations of [30 nM], [10 nM], [5 nM], [2 nM], [500 pM], [50 pM], and [5 pM] were used. Transfections were performed into 20,000-40,000 cells in 500 µl DMEM+FBS using 25 µl of DMEM without FBS and 1 µl of siPORT reagent after incubating for 20 minutes at room temperature per well. Media was replaced with fresh DMEM+FBS after 8-12 hours. Dual luciferase plasmid transfections were either performed at the same time or 24 hours later using the FuGene 6 reagent. When HiPerfect was used, the conditions followed those used for siRNA transfections. 51 Affinity purification Short version: Double stranded miR-1224-5p RNA containing a sense strand 5’ biotin modification and mismatch were purchased from IDT. Pulldown experiments were performed as previously described(Orom & Lund, 2007) with some changes. Long version: Double stranded miR-1224-5p RNA containing a sense strand 5’ biotin modification and mismatch were purchased from IDT. Streptavidin agarose beads were pre-washed, aliquoted in 500 µl lots, and stored for up to a week at -20°. Five washes with lysis buffer were performed rather than 3; after the final wash, 450 µl buffer was removed, samples were incubated for 5 minutes at 80° and quenched on ice for 2 minutes before isolating RNA. RNA isolations were performed using the RNAqueous kit (Ambion) rather than trizol extraction as described. qRT-PCR was used to observe mRNA isolation as previously described, using oligonucleotide primers specific for CCR5, Renilla luciferase, firefly luciferase; and GAPDH for normalization. 52 Electrophoretic Mobility Shift Assays Short version: A synthetic hsa-miR-1224-5p was tested for its ability to bind transcripts harboring the CCR5 -1 PRF signal (247 nt) or HIV-1 -1 PRF signal (315 nt) using a modified version of the EMSA protocol from (E. Lund, Güttinger, Calado, Dahlberg, & Kutay, 2004). Long version: Transcripts harboring the CCR5 -1 PRF signal (247 nt) or HIV-1 -1 PRF signal (315 nt) were synthesized from DNA templates using T3 RNA polymerase using MEGAscript, and purified using MEGAClear kits (Ambion). HPLC purified hsa-miR-1224-5p (5’GUGAGGACUCGGGAGGUGG3’) RNA oligonucleotide was purchased from Integrated DNA Technologies, and was 5’-[32P]-labeled by using the KinaseMax kit (Ambion). Small amounts of the CCR5 and HIV-1 derived mRNAs were also 5’-[32P]labeled and used as markers. CCR5 or HIV RNA dilutions at 2x final concentration were mixed with equal volumes of 1.0 nM 5’-[32P]-labeled hsa-miR-1224-5p RNA. Samples were incubated at 37⁰ for 30 min in HB buffer (50 mM Tris, pH 7.5, 0.1 mM EDTA, 10 mM NaCI, 10 mM MgCI2, 3% glycerol, 0.05% bromephenol blue) and immediately separated through 10% native polyacrylamide gels. For experiments with RNA refolding the RNA mix was incubated at 90⁰ for 5 sec, cooled quickly to 60⁰ and then slowly to 37⁰ (0.02⁰/sec). The electrophoresis buffer was 34 mM Tris-66 mM HEPES pH 7.5, 0.1 mM EDTA, 10 mM MgCl2. In parallel, experiments using CCR5 derived and miR-1224-5p RNA were separated through an 8% denaturing polyacrylamide gel and visualized using a phosphorimager. 53 Chemical Protection Assays Short version: Chemical protection assays were performed as described in (Christiansen, 1988) while SHAPE was performed as in (Wilkinson & Merino, 2006). Long version: Dimethylsulfate, kethoxyl and CMCT were used to probe the solvent accessibility of individual bases, while NMIA was employed to probe ribose 2’-OH groups11 in [32P]labeled run-off transcripts. In a separate experiment, synthetic CCR5 (139 nM) and hsa-miR-1224-5p (1.1 M) RNAs were annealed at 37⁰ for 30 min in 33 mM HEPES, pH 8.0, 33 mM NaCl, 10 mM MgCl2 . Structure probing with NMIA and reverse transcription reactions were subsequently performed as described11. Products were separated through 8% denaturing polyacrylamide gels, and visualized using a phosphorimager 54 In-vitro transcription for molecular tweezers Short version: Two DNA oligonucleotides were purchased from IDT: 1. 5' - TAA TAC GAC TCA CTA TAG GGG GAA AAT ATA TCA AAT CGT TCG TTG AGC G - 3' 2. 5' - GAT ACT GAC TGT ATG GAA AAT GAG AGC TGC - 3' and used pJD827 as template to PCR amplify dsDNA for in-vitro transcription using the Applied Biosystems Megascript Kit. RNA products were DNAse digested, phenol extracted, passed through a sephadex G-25 column (GE healthcare), and ethanol precipitated. Long version: 1. Oligonucleotides were chosen to anneal to the sequence immediately flanking the sequence of interest as well as contain the sequence of the DNA 'handles.' 2. The Roche 2x PCR Master Mix (K0171) was used with 0.5 ul of each oligo, approximately 100 ng plasmid, water, and mix to 50 ul. 3. PCR thermocycler conditions were as follows: a. 95 C denaturing for 10 seconds. b. 50 C annealing for 30 seconds with a ramping time decreased to 0.5 C / second. c. 68 C extension for 45 seconds. Repeat 40 times. 4. PCR products were visualized in a 1% agarose gel and purified using the Omega Bio-Tek gel purification kit. The remaining DNA was ethanol precipitated. 5. 5. T7 transcription was performed using the Ambion (Applied Biosystems) Megascript kit (AM1334) as per the manufacturers instructions. In some cases incubations were performed for 10-16 hours at 37 C. 6. RNA products were DNAse treated after transcription using the provided DNAse and inactivated by phenol extraction. 7. The remaining RNA was passed through a sephadex G-25 column (GE healthcare) and ethanol preciptated. 8. RNA was quantified via OD260/280 measurements and by running through at 1% agarose, 1x MOPS gel using 0.5-1.0 ul ethidium bromide for visualization. 55 Common Lab Buffers 1. 10X TE (used everywhere) a. 880 ml H2O b. 100ml 1M Tris (pH 8.0) c. 20ml 0.5M EDTA 2. 50X TAE (agarose gels) a. 242g Tris base b. 57.1 ml glacial acetic acid c. 100ml 0.5M EDTA pH 8.0 d. H2O to 1litre e. autoclave 3. 10X TBE (PAGE gels) a. 108g Tris base b. 55g Boric Acid c. 40ml 0.5M EDTA d. H2O to 1litre e. Filter sterilize ok, I like autoclaving it. 4. 10X MOPS (northern gels) a. 41.8g Morpholino sulphonic acid b. 20ml 1M NaOAC c. 20ml 0.5M EDTA d. H2O to 1litre e. Autoclave i. Opinions differ about what color it should be after autoclaving. I go with the Ambion note which says “Our MOPS running buffer has been yellow forever and it works great.” 5. 20X Sodium Boric Acid Running Buffer (fast agarose gels)[2,4] a. 8g NaOH b. 40g Boric Acid c. H2O to 1 litre. d. pH to 8.0 with Boric acid (turns out to be ~ 50g total) e. probably should autoclave it, haven’t tried yet. f. This apparently is better at separating smaller nucleic acid fragments, for larger use: 6. 20x Lithium Acetate Running Buffer a. 6.6g lithium acetate b. H2O to 1 litre. 56 References Christiansen, J. (1988). Enzymatic and Chemical Probing of Ribosomal RNA - Protein Interactions. Methods in Enzymology, 59, 456-468. Gey, G. O., Coffman, W., & Kubicek, M. T. (1952). Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer res, 12(4), 264–265. Gossen, M., & Bujard, H. (1992). Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proceedings of the National Academy of Sciences of the United States of America, 89(12), 5547-51. Grentzmann, G., INGRAM, J. A., KELLY, P. J., GESTALAND, R. F., & ATKINS, J. F. (1998). A dual-luciferase reporter system for studying recoding signals. Rna, 4(04), 479–486. Cambridge Univ Press. Ito, H., Fukuda, Y., Murata, K., & Kimura, A. (1983). Transformation of intact yeast cells treated with alkali cations. Journal of bacteriology, 153(1), 163–168. Am Soc Microbiol. Jacobs, J. L., & Dinman, J. D. (2004). Systematic analysis of bicistronic reporter assay data. Nucleic acids research, 32(20), e160. doi:10.1093/nar/gnh157 Ke, S. H., & Madison, E. L. (1997). Rapid and efficient site-directed mutagenesis by single-tube “megaprimer” PCR method. Nucleic acids research, 25(16), 3371-2. Lund, E., Güttinger, S., Calado, A., Dahlberg, J. E., & Kutay, U. (2004). Nuclear export of microRNA precursors. Science (New York, N.Y.), 303(5654), 95-8. doi:10.1126/science.1090599 Maquat, L. E., & Serin, G. (2001). Nonsense-mediated mRNA Decay: Insights into Mechanism from the Cellular Abundance of Human Upf1, Upf2, Upf3, and Upf3X Proteins. Cold Spring Harbor Symposia on Quantitative Biology, 66(1), 313-320. Cold Spring Harbor Laboratory Press. doi:10.1101/sqb.2001.66.313 Mattapallil, J. J., Douek, D. C., Hill, B., Nishimura, Y., Martin, M., & Roederer, M. (2005). Massive infection and loss of memory CD4 T cells in multiple tissues during acute SIV infection. Nature, 434(7037), 1093–1097. doi:10.1038/nature03501 Orom, U. A., & Lund, A. H. (2007). Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods (San Diego, Calif.), 43(2), 162-5. doi:10.1016/j.ymeth.2007.04.007 57 Platt, E., Wehrly, K., & Kuhmann, S. (1998). Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. Journal of Virology, 72(4), 2855. Am Soc Microbiol. Puck, T. T., Cieciura, S. J., & Robinson, A. (1958). Genetics of somatic mammalian cells. The Journal of experimental medicine, 108(6), 945. Rockefeller Univ Press. doi:10.1084/jem.108.6.945 Ribardo, D. A., Lambert, T. J., Kevin, S., Ribardo, D. A., Lambert, T. J., & Mciver, K. S. (2004). Role of Streptococcus pyogenes Two-Component Response Regulators in the Temporal Control of Mga and the Mga-Regulated Virulence Gene emm Role of Streptococcus pyogenes Two-Component Response Regulators in the Temporal Control of Mga and the Mga-Regulated Virulence Gene emm. Society. doi:10.1128/IAI.72.6.3668 Sambrook, J., & Russell, D. W. (2001). Molecular cloning: a laboratory manual (Vol. 3). Cold Spring Harbor, NY: Cold spring harbor laboratory press. Sheets, R. (2000). History and Characterization of the Vero Cell Line. the Vaccine and Related Biological Product Advisory. Su, M. C., Chang, C. T., Chu, C. H., Tsai, C. H., & Chang, K. Y. (2005). An atypical RNA pseudoknot stimulator and an upstream attenuation signal for- 1 ribosomal frameshifting of SARS coronavirus. Nucleic acids research, 33(13), 4265. Oxford Univ Press. Vila-Coro, A., Mellado, M., Martin de Ana, A., Lucas, P., Del Real, G., Mart’\inez-A, C., & Rodriguez-Frade, J. (2000). HIV-1 infection through the CCR5 receptor is blocked by receptor dimerization. Proceedings of the National Academy of Sciences, 97(7), 3388. National Acad Sciences. doi:10.1073/pnas.050457797 Wilkinson, K. A., & Merino, E. J. (2006). Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nature protocols, 1(3), 1610-1616. doi:10.1038/nprot.2006.249 Yamazaki, S., & Takeshige, K. (2008). Protein synthesis inhibitors enhance the expression of mRNAs for early inducible inflammatory genes via mRNA stabilization. Biochimica et biophysica acta, 1779(2), 108-14. doi:10.1016/j.bbagrm.2007.11.001 Ysla, R. M., Wilson, G. M., Brewer, G., & Riza, Y. M. (2008). Assays of adenylate uridylate-rich element-mediated mRNA decay in cells. Methods in enzymology, 449, 47–71. Elsevier. doi:10.1016/S0076-6879(08)02403-8 58 59