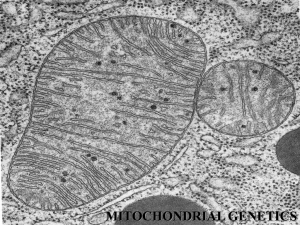
The number of mitochondrial proteins in mammals has been
advertisement

The number of mitochondrial proteins in mammals has been estimated to be about 1300. To date, pathogenic defects have been found in only a fraction of these proteins. Primary mitochondrial disease refers to disorders whose underlying genetic cause directly impairs RC composition or function. Secondary OXPHOS dysfunction, by contrast, has been described in a host of other genetic or environmental disorders, including other genetic disorders (i.e., Rett syndrome, other metabolic defects, chromosomal aneuploidies) or toxicities from drugs (i.e., valproate, statins, pesticides). The minimal prevalence of primary mitochondrial disease is one in 5000, although pathogenic mutations in mitochondrial DNA (mtDNA) may occur as frequently as one in 200 births. In the near future, developments in next generation sequencing technologies will make it possible to include high throughput sequence analysis in the diagnostic work-ups of mitochondrial patients. Often, the prediction of the pathogenicity of unknown genetic variants is not possible on the basis of sequence information alone. Usually, the level of heteroplasmy of the mtDNA variant is checked in different tissues of the patient and, in addition, in family members in the maternal lineage. Establishing a diagnosis in patients with a suspected mitochondrial disorder is often a challenge. Both knowledge of the clinical spectrum of mitochondrial disorders and the number of identified disease-causing molecular genetic defects are continuously expanding. The diagnostic examination of patients requires a multidisciplinary clinical and laboratory evaluation in which the biochemical examination of the mitochondrial functional state often plays a central role. In most cases, a muscle biopsy provides the best opportunity to examine mitochondrial function. In addition to activity measurements of individual oxidative phosphorylation enzymes, analysis of mitochondrial respiration, substrate oxidation, and ATP production rates is performed to obtain a detailed picture of the mitochondrial energy-generating system. On the basis of the compilation of clinical, biochemical, and other laboratory test results, candidate genes are selected for molecular genetic testing. In patients in whom an unknown genetic variant is identified, a compatible biochemical phenotype is often required to firmly establish the diagnosis. Metabolite analysis Prior to the biochemical examination of a muscle biopsy, metabolite analysis in blood and urine is usually performed. The results often provide important clues for the presence of a mitochondrial defect and, in some cases, can even give some indications for the location of the primary cause of the disease. Defects in the mitochondrial energy-generating system may lead to high lactate levels in blood, urine, and/ or CSF due to reduced pyruvate utilization by the mitochondria. In the case of a respiratory chain defect, the lactate/pyruvate ratio in blood will increase because of a shift in the mitochondrial redox state. Enzyme measurements Biochemical diagnostic examination of tissue and cell samples from mitochondrial patients includes measurements of enzyme activities of the oxidative phosphorylation (OXPHOS) system, consisting of complex I (EC 1.6.5.3), complex II (EC 1.3.5.1), complex III (EC 1.10.2.2), complex IV (EC 1.9.3.1), and complex V (EC 3.6.1.3) (Fig. 1). Assays to quantify OXPHOS enzyme activities are usually based on spectrophotometry. Parkinson’s disease (PD), the most common movement disorder, is characterized by age-dependent degeneration of dopaminergic neurons in the substantia nigra of the mid-brain. Non-motor symptoms of PD, however, precede the motor features caused by dysfunction of the dopaminergic system, suggesting that PD is a systemic disorder. Mitochondrial dysfunction has long been observed in PD patients and animal models, but the mechanistic link between mitochondrial dysfunction and PD pathogenesis is not well understood. Recent studies have revealed that genes associated with autosomal recessive forms of PD such as PINK1 and Parkin are directly involved in regulating mitochondrial morphology and maintenance, abnormality of which is also observed in the more common, sporadic forms of PD, although the autosomal recessive PDs lack Lewy-body pathology that is characteristic of sporadic PD. These latest findings suggest that at least some forms of PD can be characterized as a mitochondrial disorder. Whether mitochondrial dysfunction represents a unifying pathogenic mechanism of all PD cases remains a major unresolved question. Impairment of the ubiquitin–proteasome pathway can induce the accumulation of reactive oxygen species in mitochondria, with the affected mitochondria later removed by the autophagy pathway. In addition to impaired mitophagy, decreased mitochondrial biogenesis, which may be closely linked to the TORmediated protein translation pathway, is also implicated in PD pathogenesis. Thus, pathways for protein synthesis, quality control, mitochondrial maintenance, and mitochondrial dynamics are mechanistically inter-connected in the pathogenesis of PD, and represent novel targets for disease prevention and treatment. The loss of autophagy-related genes results in neurodegeneration and abnormal protein accumulation. Autophagy is a bulk lysosomal degradation pathway essential for the turnover of long-lived, misfolded or aggregated proteins, as well as damaged or excess organelles. The accumulation and aggregation of synuclein is a characteristic feature of PD. Over-expression of -synuclein is thought to impair autophagy, suggesting the presence of a cycle of impairment and accumulation. Prior studies have shown that -synuclein is degraded by chaperone-mediated autophagy. Lewy bodies observed in PD brain tissue are proteinaceous intracellular inclusions containing ubiquitin and -synuclein among many other components. The protein Parkin, mutated in the most common cause of recessive PD, may mediate the clearance of abnormal mitochondria through autophagy. Recent studies have revealed that genes associated with autosomal recessive forms of PD such as PINK1 and Parkin are directly involved in regulating mitochondrial morphology and maintenance, abnormality of which is also observed in the more common, idiopathic forms of PD. Note however that the autosomal recessive PDs lack Lewy-body pathology that is characteristic of idiopathic PD. Neurotoxins affecting humans and also used in animal models of PD: MPTP, 6-hydroxy-dopamine (6-OHDA), rotenone, and paraquat MPTP, a selective inhibitor of PD mitochondrial complex I, directed researchers’ attention to pathological roles of mitochondria in PD and raised the possibility that environmental toxins affecting mitochondria might cause PD. Other mitochondrial toxins characterized as parkinsonism-inducing reagents include 6OHDA, rotenone, and paraquat. Studies of animal models of PD induced with these toxins suggest that mitochondrial dysfunction and oxidative stress are important pathogenic mechanisms. In humans, reduced complex I activity has been reported in both post-mortem brain samples and platelets of sporadic/idiopathic PD cases. Mitochondrial Cocktails (see M.J. Falk, 2010, (J Dev Behav Pediatr 31:610 –621) Empiric “mitochondrial cocktails” are often prescribed at great expense to families but without ability to objectively monitor clinical response or adverse effects. No standard cocktail is universally used, although several vitamin and antioxidant components are commonly included. Common vitamin supplements include thiamine (B1), riboflavin (B2), ascorbate (C), or B complexes (B50 or B100). Antioxidant supplements may include a wide range of CoQ10 formulations and doses, lipoic acid, and tocopherol (vitamin E). Although CoQ10 has clear benefit for the small subset of individuals with primary Coenzyme Q deficiency, little evidence exists to suggest global benefit in all mitochondrial RC diseases. Indeed, CoQ10 has both pro- and antioxidant properties. Intermediary metabolic modifiers inconsistently recommended for RC disease patients include L-creatine for individuals with myopathy, L-carnitine for individuals with carnitine deficiency or some mitochondrial DNA cytopathies, and folinic acid for individuals with secondary folate deficiency. L-Arginine is worthy of special mention for its apparent utility in patients with mitochondrial encephalopathy, lactic acidosis, and stroke to mitigate the neurologic sequelae of metabolic stroke if administered intravenously within 30 minutes (and perhaps up to 24 hr) of acute neurologic manifestations, as well as to reduce stroke frequency and severity with chronic oral administration. Additionally, secondary CSF folate deficiency in mitochondrial disease may present with a progressive leukoencephalopathy and white matter T2 hyperintensities evident on brain and spinal MRI; this finding can be highly responsive to folinic acid, which crosses the blood–brain barrier to replenish CSF folate. The ketogenic diet is a matter of active controversy for the treatment of intractable epilepsy in RC (respiratory chain) disease, because although it provides an alternative fuel source (ketones) to bypass glycolysis, increases Complex II-dependent respiration, and has been shown to delay progression in a mouse model of mitochondrial myopathy, it has been associated with low adherence, occasional lethality, and increased morbidity and mortality in an MTERF2 mouse model having impaired mtDNA transcription. Therapeutic monitoring of many “mitochondrial cocktails” is largely relegated to subjective observations of clinical benefit and tolerance, commonly with chronic use of initial doses despite obvious symptomatic progression. A recent review of mitochondrial cocktail components’ indications, contraindications, adverse effects, and dosing regimens for pediatric and adult patients with mitochondrial disease has been published by the Mitochondrial Medicine Society. Emerging therapeutic agents for which there is promising research in animal models and/or early-stage clinical trials in humans include mitochondrial-targeted drugs to increase their bioavailability at the site at which they are needed, lipophilic antioxidants such as probucol, transcriptional modulators, and gene-therapy aimed at mitochondrial delivery of restriction endonucleases to selectively degrade mutant mtDNA and allow cell repopulation with normal mtDNA.