Progress and Challenges in Understanding the Mechanisms of
advertisement

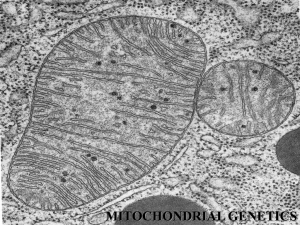
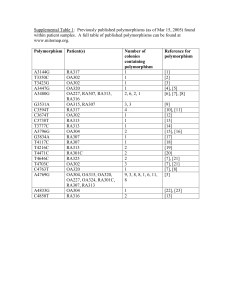
Pathogenesis of Mitochondrial Disorders Eric A. Shoubridge Montreal Neurological Institute and Department of Human Genetics McGill University, Montreal The mitochondrial oxidative phosphorylation (OXPHOS) system, responsible for aerobic energy production in nearly all cells in our body, comprises four enzyme complexes (Complexes I-IV) that make up the mitochondrial respiratory chain itself, and the ATP synthase complex (Complex V), which uses the energy generated by electron transport along the respiratory chain to produce ATP. The subunits of the five enzyme complexes are encoded in both the nuclear and mitochondrial genomes; of the 82 structural subunits, 13 are encoded in the mitochondrial genome (mtDNA), which also codes for the two rRNAs and 16 tRNAs necessary for their translation. OXPHOS deficiencies are an important cause of a wide range of neurological, neuromuscular, cardiac and endocrine disorders, and even some cancers (Koopman et al., 2012; Nunnari and Suomalainen, 2012; Schon and Przedborski, 2011; Ylikallio and Suomalainen, 2012), the minimum prevalence of which is estimated at ~1:5000 (Chinnery, 2001). The molecular basis for this extraordinary range of clinical phenotypes remains an enduring mystery. Mutations in mtDNA are the most frequent cause of mitochondrial disease in adults and more than 100 such pathogenic mutations have been identified. In the pediatric population the majority of OXPHOS disorders (~80%) are transmitted as autosomal recessive traits, usually with a severe phenotype and a fatal outcome. More than 100 nuclear genes have now been associated with OXPHOS disorders, and with the advent of whole exome sequencing that number is expanding rapidly. The genetics of mtDNA are completely different than that of nuclear genes. MtDNA is maternally inherited in mammals and it exists as a thousand copy genome. Nearly all diseasecausing mutations are heteroplasmic, meaning that both wild-type and mutant genomes are segregating in different cell types. Transmission of mutant mtDNAs from generation to generation is largely stochastic, but segregation can be rapid due to the presence of a bottleneck in germline development. This makes genetic counselling of mtDNA mutation carriers challenging. Interestingly, the majority of human pathogenic mutations occur in tRNA genes, as there appears to be a filter in development that selects against the most severe mutations in the structural genes. A biochemical, and therefore clinical, phenotype is not seen until the proportion of mutant geneomes exceeds a particular threshold. Generally, mutations in particular mtDNA genes are associated with specific and largely non-overlapping phenotypes. The molecular basis for this is not well understood. As mtDNA itself encodes for only a handful of genes the replication, maintenance, transcription, and translation of the genome is entirely dependent upon nuclear encoded, mitochondrially targeted proteins. Belying the evolutionary origins of mitochondria from proteobacteria, the enzymes responsible for mtDNA replication and transcription are homologous to those in bacteriophage. The translation machinery responsible for synthesizing the 13 mtDNA-encoded polypeptides, however, does resembles that in prokaryotes. Defects related to the handling and processing of mitochondrial mRNAs and mitochondrial translation have emerged as a important causes of OXPHOS disease, but the posttranscriptional regulation of mammalian mitochondrial gene expression is only beginning to be understood. The molecular pathogenesis of these disorders has been studied in a variety of animal (Tyynismaa and Suomalainen, 2009) models and in cells cultured from patients with mitochondrial disease. Despite the Mendelian genetics, disorders due to nuclear gene defects have even broader phenotyic variability than mtDNA related disorders. Although this is very incompletely understood, it is clear that a number of factors contribute to this including nuclear background, cell-type specific organization of the translation apparatus, and the ability of different cell types to adapt to the genetic defect. I will discuss recent progress in this area, future directions, and challenges. Chinnery, P., Turnbull, D.M. (2001). Epidemiology and treatment of mitochondrial disorders. Am J Med Genet 106, 94-101. Koopman, W.J., Willems, P.H., and Smeitink, J.A. (2012). Monogenic mitochondrial disorders. N Engl J Med 366, 1132-1141. Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145-1159. Schon, E.A., and Przedborski, S. (2011). Mitochondria: the next (neurode)generation. Neuron 70, 1033-1053. Tyynismaa, H., and Suomalainen, A. (2009). Mouse models of mitochondrial DNA defects and their relevance for human disease. Embo Rep 10, 137-143. Ylikallio, E., and Suomalainen, A. (2012). Mechanisms of mitochondrial diseases. Annals of medicine 44, 41-59.