Eq.S1.5. Compartment 5: Small intestinal wall (SV)
advertisement
")
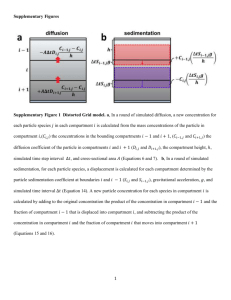
Supplement 1 for the manuscript: “Development and application of a mechanistic pharmacokinetic model for simvastatin and its active metabolite simvastatin acid using an integrated population PBPK approach” Nikolaos Tsamandouras1, Gemma Dickinson2, Yingying Guo2, Stephen Hall2, Amin Rostami-Hodjegan1,3, Aleksandra Galetin1, Leon Aarons1 1. Centre for Applied Pharmacokinetic Research, Manchester Pharmacy School, University of Manchester, Manchester, UK. 2. Eli Lilly and Company, Indianapolis, IN, USA 3. Simcyp Limited, Blades Enterprise Centre, Sheffield, UK. Contents 1. Model structure, assumptions and differential equations .................................................................... 2 2. Figures ................................................................................................................................................ 9 3. Tables ................................................................................................................................................ 11 4. References ......................................................................................................................................... 12 1 1. Model structure, assumptions and differential equations The model includes small intestinal wall, liver vascular, liver tissue, systemic blood, muscle and rest of the body (ROB) compartments for both SV and SVA. In order to account for the dissolution and absorption processes of the orally administered SV, two stomach content and two small intestinal lumen compartments referring to the solid and dissolved drug have been additionally incorporated for SV. The dissolution process was modelled according to Hintz and Johnson [1] as exemplified in Eqs.S1.1-S1.4; solid SV was treated as a powder of monodispersed spherical particles the radius of which is constant over time. It has been assumed that absorption of SV occurs only in the small intestine (no colonic absorption) and the small intestinal lumen is treated as a cylinder. SV and SVA are subject to inter-conversion and this process is modelled with the following assumptions: i) SV SVA inter-conversion in stomach and small intestinal lumen contents is negligible [2]. ii) SV to SVA hydrolysis takes place in the model compartments representing blood and tissues mediated by plasma paraoxonases and tissue carboxylesterases respectively in conjunction with chemical (non-enzymatic) hydrolysis [3]. However, the SV to SVA hydrolysis specifically in the muscle is assumed to be mediated only chemically without an enzymatic contribution. This is based on the fact that human carboxylesterases 1 (hCE1) and 2 (hCE2) are not significantly expressed in the muscle, as supported by a Northern blot analysis of adult tissue [4]. iii) SVA to SV back-transformation takes place in the liver tissue compartment mainly via an acyl-glucuronide intermediate that undergoes spontaneous cyclisation to SV at physiological pH [5, 6]. The current model neglects any possible backtransformation in the small intestinal wall due to the absence of any related in vitro information. Both SV and SVA undergo oxidative metabolism in the small intestinal wall and the liver tissue compartments, assumed to be mediated exclusively by CYP3A [7, 8]. SVA glucuronidation not accounted within lactonisation is assumed to be negligible [6]. It is also assumed that renal elimination of SV and SVA is negligible [9] and that neither SV or SVA are subject to enterohepatic recirculation [10]. The small intestinal wall is treated as a compartment where no binding (fraction unbound is 1) [11] or partition (Eq.S1.5) [12] occurs. Due to the complex nature of SV metabolism (e.g., both chemical and enzymatic hydrolysis), it has been assumed for simplicity that intestinal 2 metabolism is equally possible for both the drug entering the small intestinal wall from the small intestinal lumen or the systemic compartment. The structure of the developed model is such that it retains a physiological-mechanistic nature only in the parts which are relevant to the desired modelling purpose. As specific clinical interest is in the prediction of concentrations only in plasma, liver (efficacy) and muscle (toxicity) the developed model is much simpler compared to a typical whole body physiologically based pharmacokinetic (PBPK) model. In this way any unnecessary additional computational complexity of the system is avoided which is particularly important as the model will be subject of parameter estimation under a population pharmacokinetic modelling approach. In particular, the model rather than describing SV/SVA disposition in the several tissues which are not of clinical interest, includes a rest of body (ROB) compartment assuming no significant loss of information. It should be clearly highlighted that this compartment is not the outcome of a “proper lumping” procedure [13]. Although, we considered starting from a whole-body PBPK model and subsequently reducing it in accordance to “proper lumping” principles [13], this was not performed due to the increased complexity added by the SV to SVA conversion inside several tissues and the limited in vitro information for the quantification of this process in each of them. For example, carboxylesterases (hCE1 or hCE2) are significantly expressed in several other tissues apart from the liver and small intestinal wall (e.g. kidney, lungs, heart) [4, 14]. However, although SV to SVA conversion can be assumed to take place in all these tissues, there is no in vitro information with regard to its magnitude in order to describe this process in the different model compartments. Therefore, as the available information was not enough to build a whole-body PBPK model and to subsequently reduce it, we included an empirical rest of body compartment where SV to SVA conversion is allowed, the magnitude of which is a parameter upon estimation. As this compartment is not the outcome of a “proper lumping” procedure it should be interpreted more as an empirical peripheral eliminating compartment rather than an actual physiological space consisting of specific tissues. However, this rest of body compartment is associated with a known volume (total body volume minus the sum of volumes related to tissues explicitly described in the model) and a known blood flow (cardiac output minus the sum of 3 blood flows related to tissues explicitly described in the model). Although a tissue partition coefficient has been assigned to this rest of body compartment, it is directly estimated from the data and not from in silico mechanistic equations [15]. Hence, this estimated “rest of body” partition coefficient is not of physiological interpretation per se as it refers to an empirical peripheral compartment rather than to an actual tissue. However, the product of the estimated partition coefficient and the known volume of the “rest of body” compartment will be indicative of the extent of the compound’s distribution in this empirical peripheral space. On the contrary to the majority of the tissues where the prediction of concentration profiles is outside the scope of this model, the SV/SVA distribution in the liver tissue should be very accurately modelled. As SVA is subject of active uptake into the hepatocytes, the liver has been separated into a liver vascular and a liver tissue compartment allowing both passive diffusion and active uptake processes to be described. On the other hand the lipophilic SV is subject of perfusion- and not permeability-limited distribution into the liver. However the two-compartment liver is maintained for SV as well to allow differentiation of the SV to SVA hydrolysis rates in liver vasculature (informed by experiments in plasma) and in liver tissue (informed by experiments in liver S9 fraction). In order to satisfy perfusion-limited assumptions the permeability surface product for unbound SV influx and efflux across the hepatic basolateral membrane has been assumed to be 10,000 times greater the hepatic blood flow [12]. Finally, of specific importance is the treatment of splachnic tissues in the model structure and the related assumptions. The small intestinal wall has been preserved in the model (Manuscript Figure 1) due to its crucial importance in first pass CYP3A and hydrolysis metabolism. The rest of the splachnic tissues (spleen, large intestine, pancreas and stomach) should not be informally lumped in the ROB compartment as their physiological topology dictates that their efferent blood flows should drain in to the liver vascular compartment. This is of particular importance for a highly extracted compound as SV, because the modelling output is sensitive to whether the liver receives the correct total blood flow (sum of hepatic artery, small intestinal wall and other splachnic tissue blood flows) or not. Therefore, 4 the model presented in Supplementary Figure S1.1 was initially developed, including a “rest of splachnic tissues” compartment where the spleen, large intestine, pancreas and stomach tissues have been lumped. SV/SVA inter-conversion in this compartment was for simplicity assumed to be negligible. This compartment was the result of a “proper lumping” procedure where the tissues are originally in parallel among each other and in series connected to the liver. The lumping procedure was performed as in [13] and the volumes and blood flows of the initial non-lumped tissues were summed to derive the corresponding volume and blood flow of the lumped “rest of splachnic” compartment. The tissue partition coefficient assigned to this “rest of splachnic” compartment was predicted in silico [15] for both SV and SVA using the tissue composition parameters of the spleen. Simulations with this alternative model indicated that the exclusion of the actual “rest of splachnic” compartment from the model (as in the final model presented in Manuscript Figure 1) did not affect model predictions on all the compartments of interest (Supplementary Figure S1.2) as long as the liver receives the correct total blood flow (notice in Manuscript Figure 1 that the “rest of splachnic” blood flow is retained). The same outcome was observed when the in silico predicted “rest of splachnic” compartment partition coefficients were varied within a 3-fold error range for both SV and SVA. This indicates that even an inaccurate prediction of the partition coefficients would not have affected this observation. Therefore the actual “rest of splachnic” compartment was omitted in the final model (Manuscript Figure 1) in order to decrease the computational complexity of the differential equations system. However the blood flow efferent from these tissues (Qspl) is retained in the model to assure that the liver receives the correct total blood flow. It is acknowledged that the exclusion of the “rest of splachnic” compartment from the model is a modelling assumption and an oversimplification of reality. However, this trade-off in order to gain computational efficiency is justifiable given that the determination of SV/SVA levels in these splachnic tissues is not part of the objectives of this work and this exclusion does not affect the model predictions in the clinically relevant plasma, liver and muscle tissues. The developed mechanistic model can be described with a system of 16 ordinary differential equations. These mass balance equations for the joint SV-SVA system (Manuscript Figure 1) are 5 described below, where A(n) is the amount in the nth compartment. Abbreviations are defined in Supplementary Table S1.1. Eq.S1.1. Compartment 1: Stomach (solid SV) 𝑑𝐴(1) 3∙𝐷 𝐴(2) = −𝑘𝑔𝑒 ∙ 𝐴(1) − ∙ (𝑆𝑜𝑙𝑠𝑡𝑜𝑚 − ) ∙ 𝐴(1) 𝑑𝑡 𝜌∙𝑟∙ℎ 𝑉𝑠𝑡𝑜𝑚 Eq.S1.2. Compartment 2: Stomach (dissolved SV) 𝑑𝐴(2) 3∙𝐷 𝐴(2) = −𝑘𝑔𝑒 ∙ 𝐴(2) + ∙ (𝑆𝑜𝑙𝑠𝑡𝑜𝑚 − ) ∙ 𝐴(1) 𝑑𝑡 𝜌∙𝑟∙ℎ 𝑉𝑠𝑡𝑜𝑚 Eq.S1.3. Compartment 3: Small intestinal lumen (solid SV) 𝑑𝐴(3) 3∙𝐷 𝐴(4) = 𝑘𝑔𝑒 ∙ 𝐴(1) − 𝑘𝑠𝑖𝑡 ∙ 𝐴(3) − ∙ (𝑆𝑜𝑙𝑠𝑖𝑙 − ) ∙ 𝐴(3) 𝑑𝑡 𝜌∙𝑟∙ℎ 𝑉𝑠𝑖𝑙 Eq.S1.4. Compartment 4: Small intestinal lumen (dissolved SV) 𝑑𝐴(4) 3∙𝐷 𝐴(4) = 𝑘𝑔𝑒 ∙ 𝐴(2) − 𝑘𝑎 ∙ 𝐴(4) − 𝑘𝑠𝑖𝑡 ∙ 𝐴(4) + ∙ (𝑆𝑜𝑙𝑠𝑖𝑙 − ) ∙ 𝐴(3) 𝑑𝑡 𝜌∙𝑟∙ℎ 𝑉𝑠𝑖𝑙 Eq.S1.5. Compartment 5: Small intestinal wall (SV) 𝑑𝐴(5) 𝐴(8) 𝐴(5) 𝐴(5) = 𝑘𝑎 ∙ 𝐴(4) + 𝑄𝑠𝑖𝑤 ∙ − 𝑄𝑠𝑖𝑤 ∙ − 𝐶𝐿𝑖𝑛𝑡𝐶𝑌𝑃3𝐴,𝑠𝑖𝑤 ∙ 𝑓𝑢𝑠𝑖𝑤 ∙ 𝑑𝑡 𝑉𝑏𝑙 𝑉𝑠𝑖𝑤 𝑉𝑠𝑖𝑤 −𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑠𝑖𝑤 ∙ 𝑓𝑢𝑠𝑖𝑤 ∙ 𝐴(5) 𝑉𝑠𝑖𝑤 Eq.S1.6. Compartment 6: Liver vascular (SV) 𝑑𝐴(6) 𝐴(5) 𝐴(7) 𝐴(8) = 𝑄𝑠𝑖𝑤 ∙ + 𝑃𝑆𝑢𝑒𝑓𝑓 ∙ 𝑓𝑢𝑙𝑡 ∙ + (𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 ) ∙ 𝑑𝑡 𝑉𝑠𝑖𝑤 𝑉𝑙𝑡 𝑉𝑏𝑙 −(𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 + 𝑄𝑠𝑖𝑤 ) ∙ 𝐴(6) 𝐴(6) 𝐴(6) − 𝑃𝑆𝑢𝑖𝑛𝑓 ∙ 𝑓𝑢𝑙𝑣 ∙ − 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑙𝑣 ∙ 𝑓𝑢𝑙𝑣 ∙ 𝑉𝑙𝑣 𝑉𝑙𝑣 𝑉𝑙𝑣 Eq.S1.7. Compartment 7: Liver tissue (SV) 6 𝑑𝐴(7) 𝐴(6) 𝐴(13) 𝐴(7) = 𝑃𝑆𝑢𝑖𝑛𝑓 ∙ 𝑓𝑢𝑙𝑣 ∙ + 𝐶𝐿𝑖𝑛𝑡𝑙𝑎𝑐𝑡 ∙ 𝑓𝑢′ 𝑙𝑡 ∙ − 𝑃𝑆𝑢𝑒𝑓𝑓 ∙ 𝑓𝑢𝑙𝑡 ∙ 𝑑𝑡 𝑉𝑙𝑣 𝑉𝑙𝑡 𝑉𝑙𝑡 −𝐶𝐿𝑖𝑛𝑡𝐶𝑌𝑃3𝐴,𝑙𝑡 ∙ 𝑓𝑢𝑙𝑡 ∙ 𝐴(7) 𝐴(7) − 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑙𝑡 ∙ 𝑓𝑢𝑙𝑡 ∙ 𝑉𝑙𝑡 𝑉𝑙𝑡 Eq.S1.8. Compartment 8: Systemic blood (SV) 𝑑𝐴(8) 𝐴(6) 𝐴(10) 𝐴(9) = (𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 + 𝑄𝑠𝑖𝑤 ) ∙ + 𝑄𝑟𝑜𝑏 ∙ + 𝑄𝑚 ∙ 𝑑𝑡 𝑉𝑙𝑣 𝑉𝑟𝑜𝑏 ∙ 𝐾𝑃𝑇:𝐵,𝑟𝑜𝑏 𝑉𝑚 ∙ 𝐾𝑃𝑇:𝐵,𝑚 −𝑄𝑠𝑖𝑤 ∙ 𝐴(8) 𝐴(8) 𝐴(8) 𝐴(8) − 𝑄𝑟𝑜𝑏 ∙ − 𝑄𝑚 ∙ − (𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 ) ∙ 𝑉𝑏𝑙 𝑉𝑏𝑙 𝑉𝑏𝑙 𝑉𝑏𝑙 −𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑏𝑙 ∙ 𝑓𝑢𝑏𝑙 ∙ 𝐴(8) 𝑉𝑏𝑙 Eq.S1.9. Compartment 9: Muscle (SV) 𝑑𝐴(9) 𝐴(8) 𝐴(9) 𝐴(9) = 𝑄𝑚 ∙ − 𝑄𝑚 ∙ − 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑚 ∙ 𝑓𝑢𝑚 ∙ 𝑑𝑡 𝑉𝑏𝑙 𝑉𝑚 ∙ 𝐾𝑃𝑇:𝐵,𝑚 𝑉𝑚 Eq.S1.10. Compartment 10: Rest of body (SV) 𝑑𝐴(10) 𝐴(8) 𝐴(10) 𝐴(10) = 𝑄𝑟𝑜𝑏 ∙ − 𝑄𝑟𝑜𝑏 ∙ − 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑟𝑜𝑏 ∙ 𝑓𝑢𝑟𝑜𝑏 ∙ 𝑑𝑡 𝑉𝑏𝑙 𝑉𝑟𝑜𝑏 ∙ 𝐾𝑃𝑇:𝐵,𝑟𝑜𝑏 𝑉𝑟𝑜𝑏 Eq.S1.11. Compartment 11: Small intestinal wall (SVA) 𝑑𝐴(11) 𝐴(14) 𝐴(11) 𝐴(11) = 𝑄𝑠𝑖𝑤 ∙ − 𝑄𝑠𝑖𝑤 ∙ − 𝐶𝐿𝑖𝑛𝑡 ′ 𝐶𝑌𝑃3𝐴,𝑠𝑖𝑤 ∙ 𝑓𝑢′ 𝑠𝑖𝑤 ∙ 𝑑𝑡 𝑉𝑏𝑙 𝑉𝑠𝑖𝑤 𝑉𝑠𝑖𝑤 + 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑠𝑖𝑤 ∙ 𝑓𝑢𝑠𝑖𝑤 ∙ 𝐴(5) 𝑉𝑠𝑖𝑤 Eq.S1.12. Compartment 12: Liver vascular (SVA) 𝑑𝐴(12) 𝐴(11) 𝐴(13) 𝐴(14) = 𝑄𝑠𝑖𝑤 ∙ + 𝑃𝑆𝑢𝑑𝑖𝑓 ∙ 𝑓𝑢′ 𝑙𝑡 ∙ + (𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 ) ∙ 𝑑𝑡 𝑉𝑠𝑖𝑤 𝑉𝑙𝑡 𝑉𝑏𝑙 7 −(𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 + 𝑄𝑠𝑖𝑤 ) ∙ −𝐶𝐿𝑢𝑎𝑐𝑡 ∙ 𝑓𝑢′ 𝑙𝑣 ∙ 𝐴(12) 𝐴(12) − 𝑃𝑆𝑢𝑑𝑖𝑓 ∙ 𝑓𝑢′ 𝑙𝑣 ∙ 𝑉𝑙𝑣 𝑉𝑙𝑣 𝐴(12) 𝐴(6) + 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑙𝑣 ∙ 𝑓𝑢𝑙𝑣 ∙ 𝑉𝑙𝑣 𝑉𝑙𝑣 Eq.S1.13. Compartment 13: Liver tissue (SVA) 𝑑𝐴(13) 𝐴(12) 𝐴(12) 𝐴(13) = 𝑃𝑆𝑢𝑑𝑖𝑓 ∙ 𝑓𝑢′ 𝑙𝑣 ∙ + 𝐶𝐿𝑢𝑎𝑐𝑡 ∙ 𝑓𝑢′ 𝑙𝑣 ∙ − 𝐶𝐿𝑖𝑛𝑡𝑙𝑎𝑐𝑡 ∙ 𝑓𝑢′ 𝑙𝑡 ∙ 𝑑𝑡 𝑉𝑙𝑣 𝑉𝑙𝑣 𝑉𝑙𝑡 −𝑃𝑆𝑢𝑑𝑖𝑓 ∙ 𝑓𝑢′ 𝑙𝑡 ∙ 𝐴(13) 𝐴(13) 𝐴(7) − 𝐶𝐿𝑖𝑛𝑡′𝐶𝑌𝑃3𝐴,𝑙𝑡 ∙ 𝑓𝑢′𝑙𝑡 ∙ + 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑙𝑡 ∙ 𝑓𝑢𝑙𝑡 ∙ 𝑉𝑙𝑡 𝑉𝑙𝑡 𝑉𝑙𝑡 Eq.S1.14. Compartment 14: Systemic blood (SVA) 𝑑𝐴(14) 𝐴(12) 𝐴(16) 𝐴(15) = (𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 + 𝑄𝑠𝑖𝑤 ) ∙ + 𝑄𝑟𝑜𝑏 ∙ + 𝑄 ∙ 𝑚 𝑑𝑡 𝑉𝑙𝑣 𝑉𝑟𝑜𝑏 ∙ 𝐾𝑃′ 𝑇:𝐵,𝑟𝑜𝑏 𝑉𝑚 ∙ 𝐾𝑃′ 𝑇:𝐵,𝑚 −𝑄𝑠𝑖𝑤 ∙ 𝐴(14) 𝐴(14) 𝐴(14) 𝐴(14) − 𝑄𝑟𝑜𝑏 ∙ − 𝑄𝑚 ∙ − (𝑄ℎ𝑎 + 𝑄𝑠𝑝𝑙 ) ∙ 𝑉𝑏𝑙 𝑉𝑏𝑙 𝑉𝑏𝑙 𝑉𝑏𝑙 +𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑏𝑙 ∙ 𝑓𝑢𝑏𝑙 ∙ 𝐴(8) 𝑉𝑏𝑙 Eq.S1.15. Compartment 15: Muscle (SVA) 𝑑𝐴(15) 𝐴(14) 𝐴(15) 𝐴(9) = 𝑄𝑚 ∙ − 𝑄𝑚 ∙ + 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑚 ∙ 𝑓𝑢𝑚 ∙ 𝑑𝑡 𝑉𝑏𝑙 𝑉𝑚 ∙ 𝐾𝑃′ 𝑇:𝐵,𝑚 𝑉𝑚 Eq.S1.16. Compartment 16: Rest of body (SVA) 𝑑𝐴(16) 𝐴(14) 𝐴(16) 𝐴(10) = 𝑄𝑟𝑜𝑏 ∙ − 𝑄𝑟𝑜𝑏 ∙ + 𝐶𝐿𝑖𝑛𝑡ℎ𝑦𝑑𝑟,𝑟𝑜𝑏 ∙ 𝑓𝑢𝑟𝑜𝑏 ∙ 𝑑𝑡 𝑉𝑏𝑙 𝑉𝑟𝑜𝑏 ∙ 𝐾𝑃′ 𝑇:𝐵,𝑟𝑜𝑏 𝑉𝑟𝑜𝑏 8 2. Figures Figure S1.1: Alternative SV/SVA mechanistic model that includes an additional lumped “Rest of Splachnic” compartment. Abbreviations are defined in Table S1.1. 9 (a) (b) Figure S1.2: Model simulated concentration profiles of SV (a) and SVA (b) in plasma, liver and muscle tissues. Simulations with the full model (Supplementary Figure S1.1) that includes the “Rest of Splachnic” compartment are represented by the thick red lines. Simulations with the reduced final model (Manuscript Figure 1) where the “Rest of Splachnic” compartment has been omitted are represented by the blue dashed lines. Simulated profiles are practically identical. 10 3. Tables Table S1.1: Nomenclature in model schematic representation and mass balance equations General CLintCYP3A,i Intrinsic clearance for CYP3A mediated oxidative metabolism in compartment (i) CLinthydr,i Intrinsic clearance for hydrolysis in compartment (i) CLintlact Intrinsic clearance for lactonisation in liver tissue CLuact Active uptake clearance for unbound SVA across hepatic basolateral membrane D Diffusion coefficient fui Fraction unbound in compartment (i) h Diffusion layer thickness ka Absorption rate constant from the intestinal lumen into the epithelium kdsil Dissolution rate constant in small intestinal lumen kdstom Dissolution rate constant in stomach contents kge Gastric emptying rate constant KPT:B,i Tissue to blood partition coefficient in compartment (i) ksit Small intestinal transit rate constant PSudif Passive diffusion clearance across the basolateral membrane for unbound SVA PSueff Permeability surface product for unbound SV efflux across basolateral membrane PSuinf Permeability surface product for unbound SV influx across basolateral membrane Qha Hepatic artery blood flow Qlv Blood flow that exits the liver vascular compartment (= Qsiw+ Qha+ Qspl ) Qm Muscle blood flow Qrob “Rest of the body” compartment blood flow Qsiw Small intestinal wall blood flow Qspl Splachnic organs blood flow excluding small intestinal wall r Particle radius SI lumen Small intestinal lumen compartment SI wall Small intestinal wall compartment Soli Solubility in compartment (i) Stom Stomach contents compartment Vi Volume of compartment (i) ρ Particle density Subscripts (i) bl Systemic blood compartment lt Liver tissue compartment lv Liver vascular compartment m Muscle compartment rob “Rest of body” compartment sil Small intestinal lumen compartment siw Small intestinal wall compartment stom Stomach content compartment When any of the above abbreviations is relevant to both SV and SVA, the abbreviation referring to SVA is followed by a prime. 11 4. References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. Hintz RJ, Johnson KC. The effect of particle size distribution on dissolution rate and oral absorption. Int J Pharm. 1989;51(1):9-17. Kaufman MJ. Rate and equilibrium constants for acid-catalyzed lactone hydrolysis of HMGCoA reductase inhibitors. Int J Pharm. 1990;66(1-3):97-106. Prueksaritanont T, Qiu Y, Mu L, Michel K, Brunner J, Richards KM, et al. Interconversion pharmacokinetics of simvastatin and its hydroxy acid in dogs: Effects of gemfibrozil. Pharm Res. 2005;22(7):1101-1109. Satoh T, Taylor P, Bosron WF, Sanghani SP, Hosokawa M, Du BNL. Current progress on esterases: From molecular structure to function. Drug Metab Dispos. 2002;30(5):488-493. Prueksaritanont T, Subramanian R, Fang X, Ma B, Qiu Y, Lin JH, et al. Glucuronidation of statins in animals and humans: A novel mechanism of statin lactonization. Drug Metab Dispos. 2002;30(5):505-512. Prueksaritanont T, Tang C, Qiu Y, Mu L, Subramanian R, Lin JH. Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab Dispos. 2002;30(11):1280-1287. Prueksaritanont T, Gorham LM, Ma B, Liu L, Yu X, Zhao JJ, et al. In vitro metabolism of simvastatin in humans [SBT]identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab Dispos. 1997;25(10):1191-1199. Prueksaritanont T, Ma B, Yu N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br J Clin Pharmacol. 2003;56(1):120-124. Mauro VF. Clinical pharmacokinetics and practical applications of simvastatin. Clin Pharmacokinet. 1993;24(3):195-202. Cheng H, Schwartz MS, Vickers S, Gilbert JD, Amin RD, Depuy B, et al. Metabolic disposition of simvastatin in patients with T-tube drainage. Drug Metab Dispos. 1994;22(1):139-142. Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8(7):676-684. Gertz M, Houston JB, Galetin A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab Dispos. 2011;39(9):1633-1642. Nestorov IA, Aarons LJ, Arundel PA, Rowland M. Lumping of whole-body physiologically based pharmacokinetic models. J Pharmacokinet Biopharm. 1998;26(1):21-46. Hosokawa M. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules. 2008;13(2):412-431. Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238-1257. 12