Cytochrome P450 in Neurological Disease
advertisement

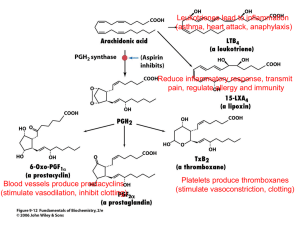
Current Drug Metabolism, 2004, 5, 225-234 225 Cytochrome P450 in Neurological Disease M. Liu, P.D. Hurn and N.J. Alkayed* Department of Anesthesiology & Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21287, USA and Department of Anesthesiology & Peri-Operative Medicine, Oregon Health & Science University, Portland, Oregon 97239, USA Abstract : Advances in a multitude of disciplines support an emerging role for cytochrome P450 enzymes and their metabolic substrates and end-products in the pathogenesis and treatment of central nervous system disorders, including acute cerebrovascular injury, such as stroke, chronic neurodegenerative disease, such as Alzheimer's and Parkinson's disease, as well as epilepsy, multiple sclerosis and psychiatric disorders, including anxiety and depression. The neural tissue contains its own unique set of P450 genes that are regulated in a manner that is distinct from their molecular regulation in peripheral tissue. Furthermore, brain P450s catalyze the formation of important brain signaling molecules, such as neurosteroids and eicosanoids, and metabolize substrates as diverse as vitamins A and D, cholesterol, bile acids, as well as centrally acting drugs, anesthetics and environmental neurotoxins. These unique characteristics allow this family of proteins and their metabolites to perform such vital functions in brain as neurotrophic support, neuroprotection, control of cerebral blood flow, temperature control, neuropeptide release, maintenance of brain cholesterol homoeostasis, elimination of retinoids from CNS, regulation of neurotransmitter levels and other functions important in brain physiology, development and disease. Key Words: Cytochrome P450, eicosanoids, EETs, neurosteroids, estrogen, cerebral ischemia, preconditioning, neuroprotection. INTRODUCTION Cytochrome P450 enzymes (P450) are heme-containing family of proteins [1] that catalyze the formation of biologically important lipid signaling molecules, including eicosanoids and steroid hormones, and play important roles in cellular adaptation to oxidative stress [2] and environ- mental toxins. The P450 enzymes are membrane-bound proteins that can be classified depending on cellular localiza- tion into microsomal P450s, which reside in the endoplasmic reticulum, and mitochondrial P450s, which are associated with the inner mitochondrial membrane. Microsomal P450s use molecular oxygen to oxidize substrates by transferring one atom of oxygen to their substrate and the other to water. This action requires electron transfer from nicotinamide adenine dinucleotide phosphate (NADPH) via two co- factors, the flavoprotein cofactor P450-reductase and cytochrome b5. Mitochondrial P450s require two additional cofactors, adrenodoxin and adrenodoxin reductase, also referred to as ferredoxin and ferrodoxin reductase. The highest concentrations of P450s are in liver, adrenals, gonads and placenta. Microsomal P450s in the liver are primarily involved in drug metabolism and detoxification of foreign chemicals (xenobiotics). P450s in adrenals, gonads, and placenta are primarily involved in steroid hormone synthesis. The last decade witnessed major advancements in the genetics and molecular biology of cytochrome P450, which *Address correspondence to this author at the Department of Anesthesiology & Peri-Operative Medicine, Oregon Health & Science University, 3181 S.W. Sam Jackson Park Rd., UHS-2, Portland, Oregon 97239-3098, USA; Tel: (503) 418-5502; Fax: (503) 494-4588; E-mail: alkayedn@ohsu.edu 1389-2002/04 $45.00+.00 allowed the identification of new members of the P450 superfamily and the assignment of P450-catalyzed biochemical reactions to specific genes. These advances have also lead to the development of gene-specific molecular tools and sensitive assays that expanded our knowledge of tissue distribution of P450s and transformed our understanding of the physiological function of P450 proteins and their role in disease state [3]. One of the most significant advances in the P450 field was the discovery that P450 enzymes are expressed in brain parenchymal cells and cerebral blood vessels and that many P450 products are endogenous constituents of brain and play important roles in brain function and neurological disease. The overall level of P450 enzymes in brain is reported to be between 0.5% and 10% [4, 5] of their level in liver. However, levels in specific neuronal populations may be equivalent or even higher than corresponding levels in hepatocytes [6]. P450 enzymes in brain are capable of sustaining some of the same activities as those described in liver, adrenals and gonads, such as drug metabolism and steroidogenesis. However, these reactions play different roles in the central nervous system (CNS) compared to peripheral tissue. Hepatic P450 enzymes metabolize and impact overall drug levels in plasma, and steroid hormones formed in gonads, adrenals and placenta are secreted into the circulation and affect distant targets in an endocrine fashion. In contrast, P450 products generated in brain remain contained within the brain and specifically impact brain function. For example, drug metabolizing P450s in brain specifically metabolize centrally acting drugs, and steroids formed in brain remain and act in brain in a paracrine manner. Furthermore, as will be reviewed below, brain © 2004 Bentham Science Publishers Ltd. 226 Current Drug Metabolism, 2004, Vol. 5, No. 3 P450s perform functions that are unique to brain such as neuroprotection, control of cerebral blood flow, formation of neuroactive steroids, temperature control [7], neuropeptide release [8], maintenance of brain cholesterol homoeostasis, elimination of retinoids from CNS, regulation of neurotransmitter levels and other functions important in brain physiology, development and disease. Finally, P450 genes are regulated differently in CNS as compared to peripheral tissue. For example, environmental toxins such as polycyclic aromatic hydrocarbons are strong inducers of P450 isoforms 1A1 and 1B1 in liver, but little induction is observed in brain [4]. Similarly, antiepileptic drug phenobarbital induces P450 2B, and steroids induce P450 3A in liver, but fail to do so in brain [4]. Finally, some P450 enzymes are sexually dimorphic in liver, but not in brain. For example, P450 2C11 is male-specific in liver, but is expressed in both male and female brains [9]. Cytochrome P450 genes (CYP) are classified into families and subfamilies based on sequence homology. In humans, there are currently 57 known P450 genes arranged into 18 families [3]. P450 isozymes belonging to families 1-4 (CYP1-4) are microsomal enzymes involved in drug and arachidonic acid metabolism. Most of these isoforms have been detected in brain, including P450 1A, 2B, 2C, 2D, 2E, 3A, 4A and 4F [4, 5]. Other P450 isoforms involved in cholesterol metabolism and steroidogenesis have also been detected in brain, including P450 7B, P450 11A1, P450 11B, P450 26A and B, P450 17, P450 21 and P450 46 [4, 5]. Below, we summarize recent studies implicating P450s in brain function and disease. Studies are grouped by P450 substrates or products, rather than specific P450 genes or reactions since, as will be explained below, the formation Liu et al. and metabolism of some of these products require multiple P450s, as well as non-P450 enzymes. NEUROSTEROIDOGENESIS Neurosteroidogenesis refers to steroid hormone synthesis in brain [10]. As illustrated in Fig. (1), all five steroid hormone classes: progestagens, glucocorticoids, mineralocorticoids, androgens and estrogens, are derived from cholesterol by the sequential action of steroidogenic enzymes. Fig. (1) highlights the crucial role of P450 enzymes in neurosteroidogenesis. The first and rate-limiting step of steroidogenesis is the conversion of cholesterol to pregnenolone in mitochondria catalyzed by cytochrome P450 side-chain cleavag, which is encoded by CYP11A1. Progesterone is formed from pregnenolone via the action of non-P450 enzyme 3hydroxysteroid dehydrogenase. Both progestagens undergo sequential 17-hydroxylation and conversion to androstenedione and dehydroepiandrostenedeione (DHEA) by microsomal P45017A1. Androstenedione is converted to testosterone via the action of non-P450 17 hydroxysteroid dehydrogenase (17HSD, also referred to as 17-ketosteroid reductase, and testosterone is aromatized to 17-estradiol via microsomal P45019A1. Progestagens can also be converted to the glucocorticoids corticosterone and cortisol via the sequential actions of 21-hydroxylase (P450 21A2) and 11hydroxylase (P45011B1). The mineralocorticoid aldosterone is formed from corticosterone via the additional action of aldosterone synthase (P450 11B2). Similar to P450 11A1, P45011B1 and P45011B2 are mitochondrial enzymes. All steroids are degraded and cleared via hydroxylation primarily by microsomal P450 enzymes of families 1-3. Below, we briefly summarize studies implicating specific Fig. (1). Role of P450 enzymes in steroid synthesis and metabolism in brain. Steroid hormones and bile acids are derived from cholesterol by the sequential action of steroidogenic enzymes, including cytochrome P450 enzymes, which are also involved in steroid metabolism and clearance from brain. Modified from [4]. Cytochrome P450 in Neurological Disease neurosteroids in brain function and disease, with special emphasis on cerebrovascular physiology and disease. Estrogens The role of estrogen in neuroprotection is well established [11]. Our previous work demonstrated that young adult female rats sustain smaller infarcts after experimental stroke induced by middle cerebral artery (MCA) occlusion compared to age-matched males [12]. This sex difference in ischemic brain injury in young adult rats disappeared after surgical ovariectomy and was absent in middle-aged, reproductively senescent rats [13], presumably due to loss of ovarian function. We subsequently demonstrated that estradiol replacement in ovariectomized [14] and reproductively senescent female rats [13] restores the protection observed in young adult females against cerebral ischemia, suggesting that estradiol is neuroprotective when administered at physiologically relevant concentrations. The mechanism of protection by estradiol is in part mediated via upregulation of neuroprotective genes, such as bcl-2, in neurons within the peri-infarct region [15]. To examine the effect of local tissue estrogen formation on ischemic brain injury, mice with targeted deletion of P450 aromatase (CYP19A1), which are incapable of converting testosterone to estrogen, were subjected to MCA occlusion and brain injury was compared to wild type with intact tissue aromatase. Mice with targeted disruption of exon 9 of the CYP 19A1 gene (aromatase knock-out mice) sustained increased brain injury after experimental stroke compared to wild-type mice with intact P450 aromatase gene, suggesting an important role for P450 aromatase in protection from ischemic brain injury [16]. Estrogens are metabolized into catechol estrogens, 2- and 4hydroxy estrogens, via the actions of P450 1A and 1B. Both genes are expressed in brain, [17] and catechol estrogens have been detected in normal brain [18] and implicated in neuronal cell death [19]. Androgens As mentioned above, testosterone is produced in brain from DHEA, which in turn is produced from pregnenolone via the action of CYP17A1. Cytochrome P450 7B is the major P450 isoform involved in terminal metabolism and elimination of progesterone and androgens in brain. In humans, DHEA and DHEA sulfate (DHEAS) are the most abundant circulating plasma steroids during development and puberty, and DHEA and its precursor pregnenolone are detected in brain at higher levels than in plasma [5]. Their levels decline with aging and stress, including stress related to acute and chronic illness. Age- and stress-related decline in DHEA levels prompted studies on the role of these compounds in age-related decline in memory and cognitive abilities [20] and protection from neurodegenerative disease [21] and cerebral ischemia. In support of a trophic function, DHEA and DHEAS increase neurite outgrowth and synapse formation in the developing brain [10], promote neuronal survival and differentiation [22], protect hippocampal neurons against neurotoxicity induced by the glutamate agonists both in vivo and in vitro [23] and improve recovery of function after traumatic brain injury in rats [24]. These effects may in part be mediated via N-methyl-D-aspartate (NMDA) receptor. For example, the effects of pregnenolone and DHEAS to Current Drug Metabolism, 2004, Vol. 5, No. 3 227 enhance spatial memory in mice is thought to result from their positive modulatory action at the NMDA receptor [10]. Finally, DHEAS, as well as progesterone and pregnenolone sulfate, bind sigma receptors with high affinity. Sigma receptors are membrane-associated intracellular protein receptors with unknown function. The sigma-1 receptor is associated with endoplasmic reticulum membrane and is mainly expressed in the central nervous system, but it can also be found in steroidogenic tissue, such as the gonads and adrenal gland, suggesting that it may be involved in steroid metabolism or action. The natural ligand for sigma receptors is unknown. However, a wide range of structurally diverse chemicals has been shown to bind these receptors, most notably neurosteroids. This action may be important to neurological function and disease since the ability of DHEAS to improve memory has been attributed to its action on sigma receptors, and synthetic sigma-1 receptor ligand 4phenyl-1-(4-phenylbutyl) piperidine is protective against cerebral ischemia, presumably by suppressing nitric oxide release in response to NMDA [25]. Progesterone We previously demonstrated that chronic progesterone administration reduces cortical infarct in middle-aged, reproductively senescent females, whose ovarian function has naturally abated, and that the protection is not related to effects of progesterone on cerebral blood flow [13]. Similarly, progesterone administration during the reperfusion period reduces ischemic injury in ovariectomized, young adult female rats [26]. Progesterone has also been shown to facilitate cognitive recovery after traumatic brain injury, presumably by reducing edema and secondary neuronal loss [27]. Finally, progesterone promotes myelination of injured nerves, likely by promoting oligodendrocyte differentiation [10]. In peripheral nerves, progesterone is produced from pregnenolone by Schwann cells, and blocking its local synthesis or action after cryolesioning of the male mouse sciatic nerve impairs remyelination of the regenerating axons, whereas administration of progesterone to the lesion site promotes the formation of new myelin sheaths [28]. In addition to these trophic effects, progesterone has also been shown to exhibit acute effects. Progesterone reduces membrane lipid peroxidation after traumatic brain injury, likely through membrane-stabilizing antioxidant effect [29]. Progesterone is also known to modulate-aminobutyric acid (GABA) receptor channel activity and expression [30] and attenuate excitatory neuronal responses [31], which may underlie progesterone's anxiolytic [32] and antiepileptic [33] properties. A synthetic analogue of pregnanolone sulfate inhibits NMDA-induced currents and cell death in primary cultures of rat hippocampal neurons [34]. Progesterone metabolites allopregnanolone and pregnenolone are endogenous agonists of GABA(A) receptor. GABA is the major inhibitory neurotransmitter in the mammalian CNS, and rapid synaptic inhibition is mediated through activation of GABA (A) receptors. Upon administration, these steroids exhibit anesthetic, sedative and anxiolytic actions, which may have implications in CNS disorders as diverse as epilepsy [35], anxiety [36], insomnia [37], migraine and cluster headache [38]. Consistent with a memory-enhancing function, pregnenolone sulfate injection into the amygdala enhanced 228 Current Drug Metabolism, 2004, Vol. 5, No. 3 post-training memory processes [39]. Depending on subunit composition, anchoring or regulatory proteins, and posttranslational modifications, neurosteroids act on GABA(A) receptor to elicit either agonistic or antagonistic activity [40]. For example, pregnenolone sulfate is an excitotoxin that has been shown to exacerbate NMDA-induced death of hippocampal neurons [41] and retina [42], presumably via cytochrome c release and caspase activation [43]. Corticosteroids To examine the effect of psychological stress on stroke outcome, we previously modeled social stress by exposing mice repeatedly to an aggressive male mouse before induction of a controlled ischemic insult [44]. Stressed mice sustained larger infarcts after cerebral ischemia compared to unstressed mice, which was associated with lower expression of bcl-2 mRNA. In addition, social stress had no effect on infarct size in transgenic mice that constitutively express increased neuronal bcl-2, suggesting that stress may exacerbate injury by suppressing bcl-2 expression. In this study, we measured postischemic concentrations of plasma corticosterone, the major adrenal gland-derived stress hormone in mice, which correlated with larger infarcts in wild-type mice. However, as illustrated in Fig. (1), glucocorticoids are also produced endogenously in brain, which may play important roles in psychiatric disorders and neurodegenerative disease. For example, stress increases brain concentrations of the neurosteroid allotetrahydrodeoxycorticosterone (THDOC), an allosteric modulator of GABA(A) receptor, which has been implicated in epilepsy, post-traumatic stress disorder and depression [45]. THDOC has also been shown to attenuate the behavioral and neuroendocrine consequences of repeated maternal separation during early life [46], which may have negative consequences on neuronal vulnerability and survival. CHOLESTEROL METABOLITES, BILE ACIDS AND VITAMINS D Unlike peripheral tissue, where excess cholesterol is cleared via lipoprotein particles, the blood brain barrier prevents cholesterol removal from brain via this mechanism. In brain, cholesterol is removed after hydroxylation to 24Rand 24S-hydroxycholesterol via cholesterol 24-hydroxylase (P45046A1) [47]. CYP46A1 is expressed at 100-fold higher levels in brain than in liver [47]. Elimination of cholesterol products is an important protective mechanism since these products possess neurotoxic effects and have been implicated in the pathogenesis of Alzheimer's disease [48]. Deletion of cholesterol 24-hydroxylase did not alter brain growth or myelination, but reduced sterol excretion from the CNS [49]. Cytochrome P450 46A1 is also part of the bile acid synthetic pathway. Bile salts are polar derivatives of cholesterol, formed via the action of P450 enzymes. Tauroursodeoxycholic acid (TUDCA), a hydrophilic bile acid, is protective against ischemic [50] and hemorrhagic [51] stroke, likely via inhibition of mitochondrial dysfunction and subsequent caspase activation. TUDCA also improves survival and function of nigral transplants in a rat model of Parkinson's disease [52] and reduces striatal neuropathology in a transgenic animal model of Huntington's disease [53]. Liu et al. Cholesterol is also the precursor of vitamin D, which plays an important role in normal bone growth, calcium and phosphorus metabolism, and tissue differentiation. Synthesis of active vitamin D (1,25-dihydroxycholecalciferol, calcitriol) from its endogenous precursor, 25-hydroxyvitamin D3 (25OHD3), is catalyzed by P450 27B1 (25-OHD3 1-hydroxylase), a mitochondrial P450 enzyme inducible by parathyroid hormone [54]. A mutation in CYP27B1 gene is associated with Vitamin D-dependent rickets type I [3]. Because P45027B1 [55] is expressed in brain, and Vitamin D receptors are expressed in both brain and immune cells, Vitamin D may have an immune modulator function and may play a role in autoimmune disease [56]. In agreement with this idea, Vitamin D administration has been shown to block the progression of relapsing encephalomyelitis in animal models of multiple sclerosis [57]. CYP27A1 has been suggested to be an anti-atherogenic enzyme because of its important role in converting cholesterol into polar metabolites [58]. A genetic defect in P45027A1 leads to the neurological disorder cerebrotendinous xanthomatosis, a rare autosomal recessive lipid-storage inherited disease characterized by abnormal bile synthesis and deposition of cholesterol and its 5-reduced derivative cholestanol in various tissues, including neural tissue, leading to progressive CNS neuropathy marked by dementia, spinal cord dysfunction, and cerebellar ataxia [59]. Finally, vitamin A (retinoid) metabolite retinoic acid (RA) is degraded by P45026A and 26B. Genetic linkage analysis maps Alzheimer's disease to genetic loci containing P450 genes involved in RA degradation [60]. Furthermore, deletion of P450 reductase, an electron donor to all microsomal P450s, in transgenic mice is associated with severe inhibition of vasculogenesis, hematopoiesis and severe developmental defects in brain, which was associated with elevated levels of retinoic acid and reduced levels of retinal. The phenotype was partially reversed by limiting exposure to retinoic acid [61]. DRUG METABOLISM, TOXINS AND ANESTHETICS Cytochrome P450 enzymes are highly inducible in response to environmental toxins, and play important roles in drug metabolism and bioactivation, foreign chemical detoxification and cellular adaptation to oxidative stress [2, 62]. However, as mentioned above, brain P450s play specific roles in local metabolism and inter-individual variations in responses to centrally acting drugs, alcohol, toxins and anesthetics [63]. Furthermore, brain and liver P450s respond differently to the same inducers, likely reflecting differences in molecular regulation. For example, the classical inducers of liver P450 enzymes,-naphtoflavone and phenobarbital, have little effect on P450 levels in brain [4]. On the other hand, brain P450 levels are affected by such solvents as ethanol and toluene as well as nicotine, antiepileptic drug phenytoin, neuroleptic drugs clozapine and sulpiride, and antidepressant drug mianserin [4-6]. Many drug-metabolizing P450s are expressed at the blood-brain interface and in brain regions not protected by the blood brain barrier, such as the choroid plexus, the median eminence, area postrema and the posterior pituitary. This lead to the notion that the presence of P450 in these locations represent an 'enzymatic Cytochrome P450 in Neurological Disease barrier' evolved to protect the brain from toxic chemicals [64, 65]. Specific drug-metabolizing P450s deserve a special discussion because of their importance in metabolizing centrally acting drugs and toxins. P450 2E1 is inducible by ethanol and nicotine and metabolizes ethanol to acetaldehyde, a highly reactive neurotoxin [6]. This isoform is also capable of generating reactive oxygen species (ROS) that can induce oxidative stress and cytotoxicity [66] and has been implicated in the development of fetal alcohol syn- drome [67] characterized by cognitive and behavioral deficits. P450s in general represent a significant source of ROS that in peripheral tissue may contribute to ischemia reperfusion injury [68]. Finally, P450 2E1 is responsible for the metabolism of many volatile anesthetics such as halothane, isoflurane, diethyl ether and chloroform [63]. Other anesthetics, including opioids, benzodiazepines and local anesthetics are metabolized by CYP3A4 [63]. Members of the CYP3A subfamily metabolize approximately 50% of drugs in therapeutic use [6]. Nicotine can induce CYP2B1, and CYP2B6 is higher in brain regions of smokers than nonsmokers [6]. Both CYP2E1 and CYP2B6 activate tobacco smoke procarcinogens, and CYP2B6 metabolizes and activates a number of neurotoxins, such as methylenedioxymethamphetamine (MDMA, or "ecstasy"), cocaine and the insecticide methyl-parathion [6]. Finally, P450 isoforms have been implicated in the metabolism of neurotransmitters. For example, CYP2D and CYP2E enzymes are expressed in dopaminergic cells and have been implicated in dopamine metabolism [6]. Toluene and clozapine induce P450 2D4, which may protect against Toluene-induced rise in reactive oxygen species and in the pharmacological actions of clozapine. CYP2D isoforms may also play a role in Parkinson's disease. Mutations in CYP2D6 are associated with increased incidence of Parkinson's disease [69] and Alzheimer's Current Drug Metabolism, 2004, Vol. 5, No. 3 229 disease variants with [70] and without Lewy bodies [71]. Furthermore, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), the neurotoxic chemical capable of inducing Parkinson's disease, is an inhibitor of P450 2D6 [72], and P450 2D isoforms are also known to metabolize neurotoxic and Parkinson-inducing dopamine metabolite tetrahydroisoquinoline [4]. CYP2D6 also metabolizes many centrally active drugs, such as tricyclic antidepressants, selective serotonin reuptake inhibitors, neuroleptics and anticonvulsants, as well as MPTP. Expression of CYP2D6 in brain and its involvement in the metabolism of MPTP supported a role for this enzyme as a susceptibility factor for Parkinsonism. Higher expression of CYP2D6 in brain was detected in alcoholics compared to non-alcoholics, possibly explaining altered sensitivity of alcoholics to centrally acting drugs and to the mediation of neurotoxic and behavioral effects of alcohol [6]. Finally, CYP2D6 is the principal enzyme involved in MDMA metabolism [73]. ARACHIDONIC ACID METABOLISM As illustrated in Fig. (2), arachidonic acid (AA) is metabolized via 3 enzymatic pathways: cyclooxygenase (COX), lipoxygenase (LOX) and P450 enzymes into an array of biologically active eicosanoids. Arachidonic acid is metabolized by P450 epoxygenases and hydroxylases into epoxyeicosatrienoic (EETs) and hydroxyeicosatetraenoic acids (HETEs), respectively. The P450 metabolites of AA are of special interest because of their vasoactive properties and their involvement in vascular control and cerebro- vascular disease, such as subarachnoid hemorrhage (SAH) and ischemic stroke [74]. The brain and cerebral blood vessels express AAmetabolizing P450 enzymes, and both EETs and HETEs are produced in brain parenchymal tissue and blood vessels [74]. The major P450 AA metabolite produced in blood vessels is 20-hydroxyeicosatetraenoic acid (20-HETE), a potent vasoconstrictor in the cerebral circulation and a product of the P450 4A family. 20-HETE plays an important role in the Fig. (2). Enzymatic pathways of arachidonic acid metabolism. Arachidonic acid (AA) is metabolized via cyclooxygenase (COX) into prostaglandins (PG), via lipoxygenase (LOX) into leukotriens (LT) and lipoxins (LX), and via P450 epoxygenase and hydroxylase into epoxyeicosatrienoic acids (EETs) or hydroxyeicosatetraenoic acids (HETEs), respectively. 230 Current Drug Metabolism, 2004, Vol. 5, No. 3 Liu et al. mechanism underlying autoregulation of cerebral blood flow (CBF) and contributes to the vasodilator actions of nitric oxide in the cerebral circulation [74]. Recently, 20-HETE has been shown to play a role in the acute fall in CBF after SAH [75], presumably by potentiating the vasoconstrictor response of cerebral vessels to 5-hydroxytryptamine [76]. and in renal epithelial cell lines, EETs inhibit apoptosis induced by hydrogen peroxide (H 2O2) and serum deprivation [91]. EETs on the other hand are potent dilators of cerebral blood vessels, and are produced in brain by astrocytes [77] via the action of P450 epoxygenases, which insert an epoxide group across the unsaturated carbon in any one of four double bonds of AA, yielding 4 regioisomers of EETs: 5,6-EET, 8,9-EET, 11,12-EET and 14,15-EET, depending on which double bond is being replaced by an epoxide group. Arachidonic acid epoxides, or EETs, are further metabolized to less active dihydroeicosatrienoic acids via the action of soluble epoxide hydrolase [8, 78]. One of the most intriguing findings in recent years is the demonstration that certain P450 isoforms involved in AA metabolism can be induced in brain, and that AA metabolites produced by these isoforms may play a neuroprotective role against ischemic cell death. This is best illustrated by examining the effect on P450 gene expression of a neuroprotective strategy such as ischemic preconditioning (IPC), whereby mild sublethal ischemic stress confers protection against subsequent severe ischemic insult. As illustrated in Fig. (3), ischemic preconditioning upregulates a battery of defense mechanisms and switches cells to a more protected phenotype, leading to enhanced tolerance against eminent lethal ischemia. Using a rat model of focal cerebral ischemic preconditioning, we previously demonstrated that mild transient ischemia induces the expression of P450 2C11 AA epoxygenase in brain [9]. Fig. (4) illustrates the experimental procedure used to induce IPC by simulating the clinical condition of transient ischemic attack (TIA), which often precedes stroke [9]. Briefly, adult male rats were subjected to three 10-min periods of middle cerebral artery occlusions (MCAO) separated by 45-min intervals or sham surgery. Three days later, rats were subjected to 2 hours of MCAO, and infarct size was measured by 2,3,5-triphenyltetrazolium chloride (TTC) at 24 hours of reperfusion. We also examined whether ischemic tolerance is associated with higher tissue perfusion during ischemia. Measurement of laser-Doppler perfusion (LDP) during MCA occlusion on day 3 demonstrated that the relative change from baseline in LDP was not different between sham and preconditioned groups, and measurement of absolute blood flow rates within MCA EETs are endogenous constituents of brain tissue and exert a variety of physiological functions in brain, including peptide hormone release from pituitary gland [8]. We previously demonstrated that astrocytes express cytochrome P450 2C11, and that astrocyte-derived EETs play an important role in coupling neuronal activity to regional blood flow [79] and maintenance of baseline CBF [80, 81]. Accumulating evidence suggests that EETs may inhibit cell death in a variety of cell types, including brain parenchymal cells. EETs are increased during myocardial ischemia [82] and reduce ischemia-reperfusion injury in heart [83]. The mechanisms underlying the anti-ischemic effect of EETs are unknown, but a number of EETs' known properties can potentially mediate this effect. As mentioned above, EETs are vasodilators in the coronary [84, 85] and cerebral [86] circulation and have been proposed to be an endotheliumdependent hyperpolarizing factor [87]. Furthermore, EETs exert anti-thrombotic [88], anti-inflammatory [89], and antipyretic [7] effects. In endothelial cell culture, EETs directly inhibit cell death induced by hypoxia-reoxygenation [90], Effect of Ischemic Preconditioning on Infarct Size and CBF Fig. (3). The adaptive responses of brain to the deleterious effects of cerebral ischemia. Mild sublethal ischemic stress confers protection against subsequent severe ischemic insult by upregulating defense mechanisms and switching brain cells to a new phenotype that is resistant to apoptotic and necrotic cell death. Modified from [117]. Cytochrome P450 in Neurological Disease Current Drug Metabolism, 2004, Vol. 5, No. 3 231 Fig. (4). Experimental protocol for induction of focal cerebral ischemic preconditioning in brain. Tolerance to ischemic injury, such as occurs during stroke, is induced in brain via brief transient ischemic attack (TIA)-like episodes consisting of three 5-min periods of MCA occlusion, separated by 45-min intervals. Tissue infarction is measured at 22 hours of reperfusion after stroke by triphenyltetrazolium chloride (TTC), and P450 2C11 mRNA and protein expression is evaluated by RNase protection assay (RPA) and Western blotting, respectively. territory using quantitative iodoantipyrine autoradiography indicated that ischemic severity was equivalent between the two groups despite the striking difference in infarct size, suggesting that ischemic tolerance in this model is not mediated via a blood-flow enhancing mechanism acting during vascular occlusion. Cortical and striatal infarcts were reduced by 50-60% in preconditioned as compared to shampretreated group. TIA alone, without subsequent stroke, did not cause tissue infarction as determined by TTC. Effect of IPC on P450 2C11 Expression To determine if P450 2C11 expression in brain is increased by IPC, rats were sacrificed and brains removed and frozen on days 1, 2 or 3 after IPC or sham surgery. Brains were then processed for quantification of P450 2C11 mRNA and protein using RNase protection assay (RPA) and Western blotting, respectively. Induction of 2C11 mRNA was apparent by day 2 in preconditioned vs. sham animals. By day 3, mRNA levels were 2-3 times higher in ipsilateral cortex and striatum in preconditioned vs. sham. The level of 2C11 protein on days 1 and 2 after IPC was not different than sham. However, in agreement with RPA results, the level of 2C11 protein in ipsilateral hemisphere on day 3 was twofold higher in preconditioned vs. sham animals [9]. Our preliminary work suggests that the reduction in infarct size in TIA-preconditioned brain is causally linked to P450 epoxygenase upregulation. Specifically, that the protection by IPC can be prevented by P450 epoxygenase inhibition and mimicked by inhibition of EETs-metabolizing epoxide hydrolase, suggesting that the epoxygenase pathway plays an important role in mediating protection by IPC and that EETs are protective against ischemic brain injury [92]. The molecular mechanisms utilized by EETs to promote cell survival and inhibit cell death are unknown. EETs activate multiple cell survival pathways [93], including the phosphoinositide 3-kinase (PI3-K)/Akt [91], the mitogenactivated protein / extracellular signal regulated kinase [94] and the cAMP/ protein kinase A pathways [95]. The PI3K/Akt pathway is a key neuronal survival signaling pathway during brain development and neurodegenerative diseases [93, 96, 97], and phosphorylation of Akt has been shown to be increased after cerebral [98, 99] and myocardial [100] ischemia and to play an important role in mediating ischemic tolerance in brain [101] and heart [102]. In addition, EETs have other properties that can potentially mediate its neuroprotective effect. These include EETs' antioxidant [90], anti-inflammatory [89] and antipyretic [7] properties. Furthermore, EETs regulate intracellular calcium concentration [103], inhibit platelet aggregation [88] and prevent leukocyte adhesion to the vascular wall [89]. A related P450 metabolite, 16(R)-hydroxyeicosatetraenoic acid, suppresses human leukocyte activation and reduces intracranial pressure in a rabbit model of thromboembolic stroke [104]. EETs may also exert their protective effect by tapping on other mechanisms of protection operative in IPC. For example, EETs have been shown to activate ATP-sensitive K+ channels [105], which are known to contribute to IPC [106]. EETs increase cAMP [95], which may in turn induce expression of neuroprotective genes known to be induced by IPC such as bcl-2 [107]. EETs may exert neurotrophic effects via their mitogenic actions [108], such as their role in mediating epidermal growth factor signaling [109]. EETs may also mediate the preconditioning effect by regulating intracellular Ca2+ loads [103], a mechanism linked to IPC [110]. EETs and P450 enzymes reciprocally regulate the cytokines interleukin-1 [111], tumor necrosis factor [95] and the sphingomyelin/ ceramide pathway [112], all of which have been implicated in the mechanism of protection by IPC [113115]. Finally, EETs exert antioxidant activity [90], which has also been linked to IPC [116]. CONCLUSIONS The central role that P450 enzymes play in the metabolism of substrates as diverse as steroids, cholesterol, vitamins, fatty acids, as well as drugs and anesthetics makes this group 232 Current Drug Metabolism, 2004, Vol. 5, No. 3 of enzymes an important player in normal CNS physiology and pathobiology. While hepatic P450s specialize in systemic drug metabolism, and adrenal and gonadal P450 specialize in systemic steroid hormone production, brain P450 evolved as a local enzyme system involved in carrying out specific brain functions, paracrine co-ordination of various neuronal, glial and vascular activities, as well as providing trophic support and adaptation to and protection from environmental stress, including oxidative and ischemic stress. ACKNOWLEDGEMENT Studies summarized above were supported in part by NIH grants RO1 NS44313, RO1 NS33668 and PO1 HL59996. The authors would like to acknowledge the excellent editorial assistance of Robin Feidelson. ABBREVIATIONS 25-OHD3 = 25-hydroxyvitamin D3 Liu et al. TIA TTC = Transient ischemic attack = 2,3,5-triphenyltetrazolium chloride TUDCA = Tauroursodeoxycholic acid REFERENCES [1] [2] [3] [4] [5] [6] [7] AA CBF = Arachidonic acid = Cerebral blood flow [8] [9] CNS = Central nervous system [10] CO = Cyclooxygenase X = Cytochrome P450 genes CYP = Dehydroepiandrostenedeione [11] [12] Nelson, D.R.; Koymans, L.; Kamataki, T.; Stegeman, J.J.; Feyereisen, R.; Waxman, D.J.; Waterman, M.R.; Gotoh, O.; Coon, M.J.; Estabrook, R.W.; Gunsalus, I.C. and Nebert, D.W. (1996) Pharmacogenetics, 6(1), 1-42. Dalton, T.P.; Puga, A. and Shertzer, H.G. (2002) Chem. Biol. Interact., 141(1-2), 77-95. Nebert, D.W. and Russell, D.W. (2002) Lancet, 360(9340), 11551162. Hedlund, E.; Gustafsson, J.A. and Warner, M. (2001) Curr. Drug Metab., 2(3), 245-263. Strobel, H.W.; Thompson, C.M. and Antonovic, L. (2001) Curr. Drug Metab., 2(2), 199-214. Miksys, S.L. and Tyndale, R.F. (2002) J. Psychiatry Neurosci., 27(6), 406-415. Kozak, W.; Kluger, M.J.; Kozak, A.; Wachulec, M. and Dokladny, K. (2000) Am. J. Physiol. Regul. Integr. Comp. Physiol., 279(2), R455-R460. Zeldin, D.C. (2001) J. Biol. Chem., 276(39), 36059-36062. Alkayed, N.J.; Goyagi, T.; Joh, H.D.; Klaus, J.; Harder, D.R.; Traystman, R.J. and Hurn, P.D. (2002) Stroke, 33(6), 1677-1684. Compagnone, N.A. and Mellon, S.H. (2000) Front Neuroendocrinol, 21(1), 1-56. Hurn, P.D. and Macrae, I.M. (2000) J. Cereb. Blood Flow Metab., 20(4), 631-652. Alkayed, N.J.; Harukuni, I.; Kimes, A.S.; London, E.D.; Traystman, R.J. and Hurn, P.D. (1998) Stroke, 29, 159-166. DHEA DHEAS = DHEA sulfate [13] Alkayed, N.J.; Murphy, S.J.; Traystman, R.J.; Hurn, P.D. and Miller, Rusa, R.; Alkayed, N.J.; Crain, B.J.; Traystman, R.J.; Kimes, A.S.; London, E.D.; Klaus, J.A. and Hurn, P.D. (1999) Stroke, 30(8), 16651670. Alkayed, N.J.; Goto, S.; Sugo, N.; Joh, H.D.; Klaus, J.; Crain, B.J.; Bernard, O.; Traystman, R.J. and Hurn, P.D. (2001) J. Neurosci., V.M. (2000) Stroke, 31(1), 161-168. EETs GABA = Epoxyeicosatrienoic acids = Gamma-aminobutyric acid [14] HETEs = Hydroxyeicosatetraenoic acids [15] 17HSD = 17 hydroxysteroid dehydrogenase 21(19), 7543-7550. [16] McCullough, L.D.; Blizzard, K.; Simpson, E.R.; Oz, O.K. and IPC LDP = Ischemic preconditioning = Laser-Doppler perfusion [17] LOX = Lipoxygenase [18] LT = Leukotrien [19] MCA = Middle cerebral artery [20] MCAO = Middle cerebral artery occlusion [21] MDMA = Methylenedioxymethamphetamine MPTP = N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [22] NADPH = Nicotinamide adenine dinucleotide phosphate [23] NMDA P450 = N-methyl-D-aspartate = Cytochrome P450 enzyme [24] PG = Prostaglandin [25] LX = Lipoxin [26] RA = Retinoic acid [27] ROS = Reactive oxygen species RPA = RNase protection assay SAH = Subarachnoid hemorrhage sEH = Soluble epoxide hydrolase THDOC = Allotetrahydrodeoxycorticosterone [28] [29] [30] [31] Hurn, P.D. (2003) J. Neurosci., 23(25), 8701-8705. Muskhelishvili, L.; Thompson, P.A.; Kusewitt, D.F.; Wang, C. and Kadlubar, F.F. (2001) J. Histochem. Cytochem., 49(2), 229-236. Weisz, J. and Crowley, W.R. (1986) Neuroendocrinology, 43(5), 543-549. Desjardins, G.C.; Beaudet, A.; Schipper, H.M. and Brawer, J.R. (1992) Endocrinology, 131(5), 2482-2484. Racchi, M.; Balduzzi, C. and Corsini, E. (2003) CNS Drug Rev., 9(1), 21-40. Yau, J.L.; Rasmuson, S.; Andrew, R.; Graham, M.; Noble, J.; Olsson, T.; Fuchs, E.; Lathe, R. and Seckl, J.R. (2003) Neuroscience, 121(2), 307-314. Bologa, L.; Sharma, J. and Roberts, E. (1987) J. Neurosci. Res., 17(3), 225-234. Kimonides, V.G.; Khatibi, N.H.; Svendsen, C.N.; Sofroniew, M.V. and Herbert, J. (1998) Proc. Natl. Acad. Sci. USA, 95(4), 18521857. Hoffman, S.W.; Virmani, S.; Simkins, R.M. and Stein, D.G. (2003) J. Neurotrauma., 20(9), 859-870. Goyagi, T.; Goto, S.; Bhardwaj, A.; Dawson, V.L.; Hurn, P.D. and Kirsch, J.R. (2001) Stroke, 32(7), 1613-1620. Murphy, S.J.; Littleton-Kearney, M.T. and Hurn, P.D. (2002) J. Cereb. Blood Flow Metab., 22(10), 1181-1188. Roof, R.L.; Duvdevani, R.; Braswell, L. and Stein, D.G. (1994) Exp. Neurol., 129(1), 64-69. Koenig, H.L.; Schumacher, M.; Ferzaz, B.; Thi, A.N.; Ressouches, A.; Guennoun, R.; Jung-Testas, I.; Robel, P.; Akwa, Y. and Baulieu, E.E. (1995) Science, 268(5216), 1500-1503. Roof, R.L.; Hoffman, S.W. and Stein, D.G. (1997) Mol. Chem. Neuropathol., 31(1), 1-11. Smith, S.S.; Gong, Q.H.; Hsu, F.C.; Markowitz, R.S.; FfrenchMullen, J.M. and Li, X. (1998) Nature, 392(6679), 926-930. Smith, S.S. (1991) Neuroscience, 42(2), 309-320. Cytochrome P450 in Neurological Disease [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53] [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] Bitran, D.; Shiekh, M. and McLeod, M. (1995) J. Neuroendocrinol., 7(3), 171-177. Herzog, A.G. (1999) Neurology, 52(9), 1917-1918. Weaver, C.E., Jr.; Marek, P.; Park-Chung, M.; Tam, S.W. and Farb, D.H. (1997) Proc. Natl. Acad. Sci. USA, 94(19), 10450- 10454. Kaminski, R.M.; Gasior, M.; Carter, R.B. and Witkin, J.M. (2003) Eur. J. Pharmacol., 474(2-3), 217-222. Brambilla, F.; Biggio, G.; Pisu, M.G.; Bellodi, L.; Perna, G.; Bogdanovich-Djukic, V.; Purdy, R.H. and Serra, M. (2003) Psychiatry Res., 118(2), 107-116. Hamilton, N.M. (2002) Curr. Top. Med. Chem., 2(8), 887-902. Limmroth, V.; Lee, W.S. and Moskowitz, M.A. (1996) Br. J. Pharmacol., 117(1), 99-104. Flood, J.F.; Morley, J.E. and Roberts, E. (1995) Proc. Natl. Acad. Sci. USA, 92(23), 10806-10810. Lambert, J.J.; Belelli, D.; Peden, D.R.; Vardy, A.W. and Peters, J.A. (2003) Prog. Neurobiol., 71(1), 67-80. Weaver, C.E., Jr.; Wu, F.S.; Gibbs, T.T. and Farb, D.H. (1998) Brain Research, 803(1-2), 129-136. Guarneri, P.; Russo, D.; Cascio, C.; De Leo, G.; Piccoli, T.; Sciuto, V.; Piccoli, F. and Guarneri, R. (1998) J. Neurosci. Res., 54(6), 787-797. Cascio, C.; Guarneri, R.; Russo, D.; De Leo, G.; Guarneri, M.; Piccoli, F. and Guarneri, P. (2002) J. Neurochem., 83(6), 1358- 1371. DeVries, A.C.; Joh, H.D.; Bernard, O.; Hattori, K.; Hurn, P.D.; Traystman, R.J. and Alkayed, N.J. (2001) Proc. Natl. Acad. Sci. USA, 98(20), 11824-11828. Reddy, D.S. (2003) Trends Pharmacol. Sci., 24(3), 103-106. Patchev, V.K.; Montkowski, A.; Rouskova, D.; Koranyi, L.; Holsboer, F. and Almeida, O.F. (1997) J. Clin. Invest., 99(5), 962966. Lund, E.G.; Xie, C.; Kotti, T.; Turley, S.D.; Dietschy, J.M. and Russell, D.W. (2003) J. Biol. Chem., 278(25), 22980-22988. Kolsch, H.; Lutjohann, D.; von Bergmann, K. and Heun, R. (2003) J. Nutr. Health Aging, 7(1), 37-41. Xie, C.; Lund, E.G.; Turley, S.D.; Russell, D.W. and Dietschy, J.M. (2003) J. Lipid Res., 44(9), 1780-1789. Rodrigues, C.M.; Spellman, S.R.; Sola, S.; Grande, A.W.; LinehanStieers, C.; Low, W.C. and Steer, C.J. (2002) J. Cereb. Blood Flow Metab., 22(4), 463-471. Rodrigues, C.M.; Sola, S.; Nan, Z.; Castro, R.E.; Ribeiro, P.S.; Low, W.C. and Steer, C.J. (2003) Proc. Natl. Acad. Sci. USA, 100(10), 6087-6092. Duan, W.M.; Rodrigues, C.M.; Zhao, L.R.; Steer, C.J.; Low, W.C. and Rodrigures, C.M. (2002) Cell Transplant., 11(3), 195-205. Keene, C.D.; Rodrigues, C.M.; Eich, T.; Chhabra, M.S.; Steer, C.J. and Low, W.C. (2002) Proc. Natl. Acad. Sci. USA, 99(16), 1067110676. Huang, D.C.; Papavasiliou, V.; Rhim, J.S.; Horst, R.L. and Kremer, R. (2002) Mol. Cancer Res., 1(1), 56-67. Fu, G.K.; Lin, D.; Zhang, M.Y.; Bikle, D.D.; Shackleton, C.H.; Miller, W.L. and Portale, A.A. (1997) Mol. Endocrinol., 11(13), 1961-1970. Holick, M.F. (2003) J. Cell Biochem., 88(2), 296-307. Cantorna, M.T.; Hayes, C.E. and DeLuca, H.F. (1996) Proc. Natl. Acad. Sci. USA, 93(15), 7861-7864. Bel, S.; Garcia-Patos, V.; Rodriguez, L.; Selva, A.; Diaz, P.; Wolthers, B.G. and Castells, A. (2001) J. Am. Acad. Dermatol., 45(2), 292-295. Okuda, K.I. (1994) J. Lipid Res., 35(3), 361-372. Goodman, A.B. and Pardee, A.B. (2003) Proc. Natl. Acad. Sci. USA, 100(5), 2901-2905. Otto, D.M.; Henderson, C.J.; Carrie, D.; Davey, M.; Gundersen, T.E.; Blomhoff, R.; Adams, R.H.; Tickle, C. and Wolf, C.R. (2003) Mol. Cell Biol., 23(17), 6103-6116. Nebert, D.W.; Roe, A.L.; Dieter, M.Z.; Solis, W.A.; Yang, Y. and Dalton, T.P. (2000) Biochem. Pharmacol., 59(1), 65-85. Chang, G.W. and Kam, P.C. (1999) Anaesthesia, 54(1), 42-50. Ghersi-Egea, J.F.; Leninger-Muller, B.; Suleman, G.; Siest, G. and Minn, A. (1994) J. Neurochem., 62(3), 1089-1096. Ghersi-Egea, J.F.; Leininger-Muller, B.; Cecchelli, R. and Fenstermacher, J.D. (1995) Toxicol. Lett., 82-83, 645-653. Current Drug Metabolism, 2004, Vol. 5, No. 3 [66] [67] [68] [69] [70] [71] [72] [73] [74] [75] [76] [77] [78] [79] [80] [81] [82] [83] [84] [85] [86] [87] [88] [89] [90] [91] [92] [93] [94] [95] [96] [65] [97] 233 Brzezinski, M.R.; Boutelet-Bochan, H.; Person, R.E.; Fantel, A.G. and Juchau, M.R. (1999) J. Pharmacol. Exp. Ther., 289 (3), 16481653. Boutelet-Bochan, H.; Huang, Y. and Juchau, M.R. (1997) Biochem. Biophys. Res. Commun., 238(2), 443-447. Gottlieb, R.A. (2003) Arch. Biochem. Biophys., 420(2), 262-267. Smith, C.A.; Gough, A.C.; Leigh, P.N.; Summers, B.A.; Harding, A.E.; Maraganore, D.M.; Sturman, S.G.; Schapira, A.H. and Williams, A.C. (1992) Lancet, 339(8806), 1375-1377. Saitoh, T.; Xia, Y.; Chen, X.; Masliah, E.; Galasko, D.; Shults, C.; Thal, L.J.; Hansen, L.A. and Katzman, R. (1995) Ann. Neurol., 37(1), 110-112. Chen, X.; Xia, Y.; Alford, M.; DeTeresa, R.; Hansen, L.; Klauber, M.R.; Katzman, R.; Thal, L.; Masliah, E. and Saitoh, T. (1995) Ann. Neurol., 38(4), 653-658. Fonne-Pfister, R.; Bargetzi, M.J. and Meyer, U.A. (1987) Biochem. Biophys. Res. Commun., 148(3), 1144-1150. Oesterheld, J.R.; Armstrong, S.C. and Cozza, K.L. (2004) Psychosomatics, 45(1), 84-87. Roman, R.J. (2002) Physiol. Rev., 82(1), 131-185. Kehl, F.; Cambj-Sapunar, L.; Maier, K.G.; Miyata, N.; Kametani, S.; Okamoto, H.; Hudetz, A.G.; Schulte, M.L.; Zagorac, D.; Harder, D.R. and Roman, R.J. (2002) Am. J. Physiol. Heart Circ. Physiol., 282(4), H1556-H1565. Cambj-Sapunar, L.; Yu, M.; Harder, D.R. and Roman, R.J. (2003) Stroke, 34(5), 1269-1275. Alkayed, N.J.; Narayanan, J.; Gebremedhin, D.; Medhora, M.; Roman, R.J. and Harder, D.R. (1996) Stroke, 27, 971-979. Fretland, A.J. and Omiecinski, C.J. (2000) Chem. Biol. Interact., 129(1-2), 41-59. Alkayed, N.J.; Birks, E.K.; Narayanan, J.; Petrie, K.A.; KohlerCabot, A.E. and Harder, D.R. (1997) Stroke, 28, 1066-1072. Harder, D.R.; Alkayed, N.J.; Lange, A.R.; Gebremedhin, D. and Roman, R.J. (1998) Stroke, 28, 229-234. Alkayed, N.J.; Birks, E.K.; Hudetz, A.G.; Roman, R.J.; Henderson, L. and Harder, D.R. (1996) Am. J. Physiol. Heart Circ. Physiol., 271, H1541-H1546. Nithipatikom, K.; DiCamelli, R.F.; Kohler, S.; Gumina, R.J.; Falck, J.R.; Campbell, W.B. and Gross, G.J. (2001) Anal. Biochem., 292(1), 115-124. Wu, S.; Chen, W.; Murphy, E.; Gabel, S.; Tomer, K.B.; Foley, J.; Steenbergen, C.; Falck, J.R.; Moomaw, C.R. and Zeldin, D.C. (1997) J. Biol. Chem., 272(19), 12551-12559. Campbell, W.B. and Harder, D.R. (1999) Circ. Res., 84(4), 484- 488. Zhang, Y.; Oltman, C.L.; Lu, T.; Lee, H.C.; Dellsperger, K.C. and VanRollins, M. (2001) Am. J. Physiol. Heart Circ. Physiol., 280(6), H2430-H2440. Ellis, E.F.; Police, R.J.; Yancey, L.; McKinney, J.S. and Amruthesh, S.C. (1990) Am. J. Physiol. Heart Circ. Physiol., 259, H1171-H1177. Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I. and Busse, R. (1999) Nature, 401(6752), 493-497. Heizer, M.L.; McKinney, J.S. and Ellis, E.F. (1991) Stroke, 22(11), 1389-1393. Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C. and Liao, J.K. (1999) Science, 285(5431), 1276-1279. Yang, B.; Graham, L.; Dikalov, S.; Mason, R.P.; Falck, J.R.; Liao, J.K. and Zeldin, D.C. (2001) Mol. Pharmacol., 60(2), 310-320. Chen, J.K.; Capdevila, J. and Harris, R.C. (2001) Mol. Cell Biol., 21(18), 6322-6331. Alkayed, N.J.; Goyagi, T.; Peng, X.; Harder, D.R.; Hurn, P.D.; Traystman, R.J. and Koehler, R.C. (2002) Pharmacology of Cerebral Ischemia, (Krieglstein, J. Eds.) Medpharm Scientific Publishers, Stuttgart, Germany, 303-310. Yuan, J. and Yanker, B.A. (2000) Nature, 407, 802-809. Fleming, I.; Fisslthaler, B.; Michaelis, U.R.; Kiss, L.; Popp, R. and Busse, R. (2001) Pflugers Arch., 442(4), 511-518. Node, K.; Ruan, X.L.; Dai, J.; Yang, S.X.; Graham, L.; Zeldin, D.C. and Liao, J.K. (2001) J. Biol. Chem., 276(19), 15983-15989. Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R. and Greenberg, M.E. (1997) Science, 275(5300), 661-665. Brunet, A.; Datta, S.R. and Greenberg, M.E. (2001) Curr. Opin. Neurobiol., 11(3), 297-305. 234 [98] [99] [100] [101] [102] [103] [104] [105] [106] [107] Current Drug Metabolism, 2004, Vol. 5, No. 3 Ouyang, Y.B.; Tan, Y.; Comb, M.; Liu, C.L.; Martone, M.E.; Siesjo, B.K. and Hu, B.R. (1999) J. Cereb. Blood Flow Metab., 19(10), 1126-1135. Namura, S.; Nagata, I.; Kikuchi, H.; Andreucci, M. and Alessandrini, A. (2000) J. Cereb. Blood Flow Metab., 20(9), 13011305. Tong, H.; Chen, W.; Steenbergen, C. and Murphy, E. (2000) Circ. Res., 87(4), 309-315. Yano, S.; Morioka, M.; Fukunaga, K.; Kawano, T.; Hara, T.; Kai, Y.; Hamada, J.; Miyamoto, E. and Ushio, Y. (2001) J. Cereb. Blood Flow Metab., 21(4), 351-360. Baines, C.P.; Wang, L.; Cohen, M.V. and Downey, J.M. (1998) J. Mol. Cell Cardiol., 30(2), 383-392. Rzigalinski, B.A.; Willoughby, K.A.; Hoffman, S.W.; Falck, J.R. and Ellis, E.F. (1999) J. Biol. Chem., 274, 175-182. Bednar, M.M.; Gross, C.E.; Russell, S.R.; Fuller, S.P.; Ahern, T.P.; Howard, D.B.; Falck, J.R.; Reddy, K.M. and Balazy, M. (2000) Neurosurgery, 47(6), 1410-1418. Lu, T.; Hoshi, T.; Weintraub, N.L.; Spector, A.A. and Lee, H.C. (2001) J. Physiol., 537(Pt 3), 811-827. Heurteaux, C.; Lauritzen, I.; Widmann, C. and Lazdunski, M. (1995) Proc. Natl. Acad. Sci. USA, 92(10), 4666-4670. Shimizu, S.; Nagayama, T.; Jin, K.L.; Zhu, L.; Loeffert, J.E.; Watkins, S.C.; Graham, S.H. and Simon, R.P. (2001) J. Cereb. Blood Flow Metab., 21(3), 233-243. Liu et al. [108] [109] [110] [111] [112] [113] [114] [115] [116] [117] Chen, J.K.; Capdevila, J. and Harris, R.C. (2000) J. Biol. Chem., 275(18), 13789-13792. Chen, J.K.; Wang, D.W.; Falck, J.R.; Capdevila, J. and Harris, R.C. (1999) J. Biol. Chem., 274(8), 4764-4769. Ohta, S.; Furuta, S.; Matsubara, I.; Kohno, K.; Kumon, Y. and Sakaki, S. (1996) J. Cereb. Blood Flow Metab., 16(5), 915-922. Chen, J.Q.; Strom, A.; Gustafsson, J.A. and Morgan, E.T. (1995) Mol. Pharmacol., 47(5), 940-947. Nikolova-Karakashian, M.; Morgan, E.T.; Alexander, C.; Liotta, D.C. and Merrill, A.H., Jr. (1997) J. Biol. Chem., 272(30), 18718- 18724. Nawashiro, H.; Tasaki, K.; Ruetzler, C.A. and Hallenbeck, J.M. (1997) J. Cereb. Blood Flow Metab., 17(5), 483-490. Liu, J.; Ginis, I.; Spatz, M. and Hallenbeck, J.M. (2000) Am. J. Physiol Cell Physiol., 278(1), C144-C153. Ohtsuki, T.; Ruetzler, C.A.; Tasaki, K. and Hallenbeck, J.M. (1996) J. Cereb. Blood Flow Metab., 16(6), 1137-1142. Wiegand, F.; Liao, W.; Busch, C.; Castell, S.; Knapp, F.; Lindauer, U.; Megow, D.; Meisel, A.; Redetzky, A.; Ruscher, K.; Trendelenburg, G.; Victorov, I.; Riepe, M.; Diener, H.C. and Dirnagl, U. (1999) J. Cereb. Blood Flow Metab., 19(11), 1229- 1237. Fisher, M. and Ratan, R. (2003) Ann. Neurol., 53(1), 10-20. Copyright of Current Drug Metabolism is the property of Bentham Science Publishers Ltd. and its content may not be copied or emailed to multiple sites or posted to a listserv without the copyright holder's express written permission. However, users may print, download, or email articles for individual use.