NSAR Polarimeter Assay
advertisement

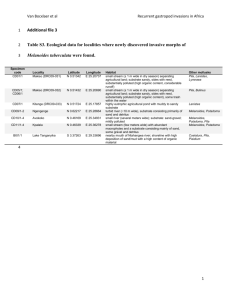
Measurement of NSAR activity on Jasco P2000 polarimeter Last Edited by Andy McMillan 12-16-2013 Making serial dilution of substrate 1. Weigh out sufficient substrate for samples. (MW = MW of amino acid + 101.08) I generally make 700 uL final volumes. Record actual weight measured. 2. Starting from 100 mM make dilutions down to needed concentrations, lowest measurable seems to be 0.125 mM for succinyl-Phe and 0.25 for succinyl-Val. Others have not be determined yet 3. Check actual concentration based on specific rotation (varies for each amino acid). Specific Rotation: Succinyl-L-Phe = 2.17 deg/M/cm Succinyl-L-Val = -0.78 deg/M/cm Succinyl-D-phenylglycine = 6.22 deg/M/cm D-amino acids should be same magnitude with opposite sign Amino acids that absorb in UV can also be checked on the UV spectrophotometer or plate reader ( is similar to free amino acid, Phe = 195@257.5, Trp = 5,579 @ 278, Tyr = 1,405@ 274.5 http://omlc.ogi.edu/spectra/PhotochemCAD/index.html) Measurement of Protein Concentration 1. Spin down some of protein stock in centrifuge in cold room to remove any aggregates 2. Measure concentration in cuvette by absorbance at 280 nm, as well as A330 to correct for light scattering Acorrected = A280- (1.928*A330) 3. Measure at several different dilutions, for typical protein expression levels I find 1:20, 1:10 and 1:5 work 4. If needed dilute with storage buffer+glycerol and repeat concentration measurement (see below for how to mix reaction) Measurement of kinetics 1. Setup buffer for 1 set of reactions for each substrate concentration in series of 2 mL eppendorf tubes (200 mM Tris, 0.1 mM MnCl2, Water needed for final volume of 1600 uL after substrate and enzyme would be added) 2. Add enzyme shortly before ready to start reaction 3. Stretching the tip of a plastic transfer pipet and cutting off the unstretched tip allows for transferring reaction mix. For metal cell a short narrow tip works best, if too long it can be bent. For the glass cell the pipet should be curved so it fits down each end of the cell, which would otherwise form an air bubble if filled quickly (only use for low specific rotation substrates that need longer pathlength). 4. Start reaction by adding 160 uL substrate, mix, then transfer to cell. Transfer into cell slowly to avoid bubbles. If a bubble is introduced it can be removed/popped by shaking the cell. 5. Measurements typically use 10s integration time, recording every 30s for 450s. Need to read at 405 nm (if need to change for some reason, filter also needs changing) 6. After each run save data, and under the File menu “Send to Analysis” to open program which can export data as a text file. Data Analysis (can be done while next reaction is running) 1. Open exported text data file in a spreadsheet 2. Fit data to from beginning of reaction to where rate begins to change The first point tends to be drastically different from actual values, so should be left out of fit b. Changing format of time column and explicitly putting in seconds value is needed for proper value of fit. c. Make sure fit uses enough decimal places, this will probably require changing to scientific notation 3. Convert rate from deg/s to mM/s using C = l =specific rotation for amino acid in units of M-1cm-1 and l = pathlength (10 cm for glass cell, 5 cm for metal). The factor of two is to account for the signal changing twice for each molecule that is converted (loss of L-amino acid, creation of D-amino acid) 4. Get kobs by dividing by enzyme concentration 5. Plot kobs vs substrate concentration in Kaleidograph a. Fit to Michelis-Menton equation (should be already defined) to determine kcat and Km b. If not reaching saturation fit low concentrations to a linear plot for kcat/Km value a.