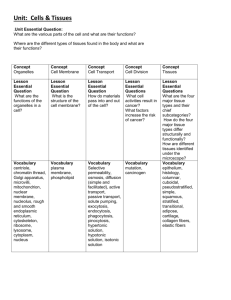
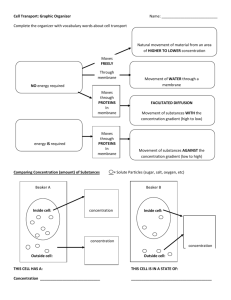
Yoo_Seung Mi_SM_201406_PhD - MacSphere
advertisement

HYDROGEL BASED MEMBRANES FOR BIOPHARMACEUTICAL AND BIOMEDICAL APPLICATIONS i HYDROGEL BASED MEMBRANES FOR BIOPHARMACEUTICAL AND BIOMEDICAL APPLICATIONS By SEUNG MI YOO, MASc. A Thesis Submitted to the School of Graduate Studies in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy McMaster University © Copyright by SEUNG MI YOO, April 2014 II DOCTOR OF PHILOSOPHY (2014) McMaster University (Chemical Engineering) Hamilton, Ontario TITLE: HYDROGEL BASED MEMBRANES FOR BIOPHARMACEUTICAL AND BIOMEDICAL APPLICATIONS AUTHOR: SEUNG MI YOO, MASc. (University of Waterloo) SUPERVISOR: Professor Raja Ghosh NUMBER OF PAGES: xxv, 141 iii Abstract Membrane technology has been actively used as a separation tool in the chemical, environmental, and biopharmaceutical industries for several decades. As membrane quality requirement in the industry has increased, efforts have been directed towards enhancement in mechanical strength, chemical durability and functionality of membranes. One of the approaches for membrane quality enhancement is based on the combination of hydrogel technology with membrane technology. This thesis focused on the application and development of hydrogel based membranes, notably hydrophilized PVDF (polyvinylidene fluoride) membrane for hydrophobic interaction membrane chromatography; the fabrication of paper-hydrogel composite membranes for membrane chromatography; development of a technique for coating alginate (a natural hydrogel) on the outer surface of a hollow fiber membrane for potential application in bioreactors and the use of hollow fiber membranes as mold for fabrication calcium alginate fibers for biomedical and tissue engineering applications. A membrane chromatography-based polishing technique was developed for removing leached protein-A and aggregates from monoclonal antibody (mAb). A commercial synthetic membrane that is known to be hydrophilized by hydrogel grafting was employed to develop this polishing process that resulted in highly pure mAb, free from aggregates and protein-A. This mAb polishing technique could easily be integrated with a hydrophobic interaction membrane chromatography based mAb purification process. iv A paper-hydrogel composite membrane was developed as an inexpensive alternative to commercial synthetic membranes used for carrying hydrophobic interaction membrane chromatography. Poly(N-vinylcaprolactam) or PVCL hydrogel was coated on Whatman filter paper to prepare these membranes. These environment responsive membrane which responded to changes in salt concentration, gave excellent fractionation of multicomponent protein mixtures. As case study, a mixture of immunoglobulin G, human serum albumin and insulin was fractionated. A technique for modifying the surface of synthetic hollow fiber membranes with alginate (a natural hydrogel) was developed. This manner of surface modification led to the improvement in membrane mass transport. The alginate was cross-linked on the outer surface of the membrane by diffusion of the cross-linker (calcium ions) through the membrane pores. The calcium alginate coating layer was characterized by optical and transmission electron microscopy, contact angle measurement, hydraulic permeability measurement and by examining solute transport. Hollow and solid calcium alginate fibers were fabricated using a novel hollow fiber membrane based moulding technique. The pore present on the hollow fiber membrane served as the reservoir for the calcium chloride solution with cross-linked the alginate within the lumen. The calcium alginate fibers produced were characterized by optical, transmission electron, and scanning electron microscopy. Cell immobilization experiments were carried out to demonstrate biocompatibility and potential for tissue engineering applications. v Acknowledgements I would like to express my sincere gratitude to all those who supported my research leading to this thesis. First, I would like to express my deepest gratitude to my research supervisor, Dr. Raja Ghosh, he has been a tremendous mentor for me. I would like to thank him for encouraging my research projects. This led me to grow more creative and developed my thinking about the project. His advice on both research, as well as on my career have been priceless. Also, I appreciate his efforts to give me the opportunities to experience worldwide conferences; it made me understand the relationship between research and industry. Without his guidance and support this dissertation would not be possible. I would also like to acknowledge my supervisory committee membranes, Dr. James Dickson, Dr. Ravi Selvaganapathy for their valuable advice and encouragement by participating in my annual committee meetings. Dr. Todd Hoare is also acknowledged for kindly allowing me access to his facilities. I would like to acknowledge the funding support from Natural Sciences and Engineering Research Council of Canada, mAbnet research funding. I also thank my personal scholarship: International Excellence Awards, and best TA awards. Also, the ambassadorial scholarship from Rotary international helped me to explore a new country with fantastic Rotarians. vi Dennis, Lu, Mukesh and Zheng taught me the proper use of experimental machinery and guided my grad student life. Bioseparation research group, Annabel, Evan, Si, Rahul, and Pedram, for their warm hearts and always providing a happy environment in the lab. Special thanks to summer students, Ryan Sungho, Steven, Simon, who made for a fun and dynamic environment while we were doing research. My dear colleagues, Scott, Matt, Emilia, Daryl, Chris, Lina, and Wein whom I thank for sharing their knowledge of different research areas and their friendship. Moreover, I would like to thank my dearest and sweetest friends Meng, Yesol, and Soomin for their support throughout my education. Dr. JungBok Lee who educated me with his biology backgrounds and cell culture practise for the last two years without any reward. I would like to give my thanks to the staff in Chemical Engineering Department, Kathy Goodram, Lynn Falkiner, Melissa Vasil, and Cathie Roberts for their assistance. Special acknowledge to Paul Gatt for fabricating various custom designed membrane modules. Finally, I would like to express my deep gratitude to my family, mom, dad, and my younger brother for all of the sacrifices that you have made on my behalf. Your prayer for me was what sustained me thus far. And my Canadian family, Kathi and Ray Smith, for without their support and belief in me, I could not have even started my graduate studies in Canada. vii Table of Contents Chapter 1. Introduction of membranes with hydrogels....................................................... 1 1.1 Membrane technology .............................................................................................. 1 1.2 Membrane chromatography ...................................................................................... 3 1.3 Hydrogel based membrane...................................................................................... 10 1.4 Thesis outline .......................................................................................................... 12 1.5 References ............................................................................................................... 16 Chapter 2. Simultaneous removal of leached protein-A and aggregates from monoclonal antibody using hydrophobic interaction membrane chromatography............................... 20 2.1 Abstract ................................................................................................................... 21 2.2 Introduction ............................................................................................................. 22 2.3 Materials and Methods ............................................................................................ 27 2.3.1 Materials .......................................................................................................... 27 2.3.2 Methods............................................................................................................ 28 2.4 Results and Discussion ............................................................................................ 29 2.5 Conclusion............................................................................................................... 41 2.6 References ............................................................................................................... 42 Chapter 3. High-resolution hydrophobic interaction chromatographic separation of multiprotein mixture using paper-based composite membranes .............................................. 47 viii 3.1 Abstract ................................................................................................................... 48 3.2 Introduction ............................................................................................................. 49 3.3 Materials and Methods ............................................................................................ 55 3.3.1 Materials .......................................................................................................... 55 3.3.2 Separation of protein mixture with HIC butyl column .................................... 56 3.3.3 Paper based composite membrane preparation ................................................ 56 3.3.4 Membrane chromatography set-up .................................................................. 57 3.3.5 Hydraulic testing of membranes ...................................................................... 58 3.3.6 SDS-PAGE ...................................................................................................... 58 3.3.7 SEC-HPLC ....................................................................................................... 58 3.4 Results and Discussions .......................................................................................... 59 3.4.1 Optimization of protein separation with resin based hydrophobic interaction chromatography ........................................................................................................ 59 3.4.2 Composite paper based membrane characterizations (salt responsive and protein binding/elution test) ...................................................................................... 63 3.4.3 Process optimization (elution mode) ............................................................... 67 3.5 Conclusions ............................................................................................................. 75 3.6 References ............................................................................................................... 77 Chapter 4. Hollow fiber membrane surface coating with natural hydrogel for bioreactor 81 ix 4.1 Abstract ................................................................................................................... 82 4.2 Introduction ............................................................................................................. 83 4.3 Materials and Methods ............................................................................................ 87 4.3.1 Materials ........................................................................................................... 87 4.3.2 Methods............................................................................................................ 88 4.4 Results and Discussions .......................................................................................... 92 4.5 Conclusions ........................................................................................................... 105 Chapter 5. Fabrication of alginate fibers using a microporous membrane based molding technique ........................................................................................................................ 111 5.1 Abstract ................................................................................................................. 112 5.2 Introduction ........................................................................................................... 113 5.3 Materials and Methods .......................................................................................... 118 5.3.1 Fabrication of calcium alginate hollow fibers ............................................... 119 5.3.2 Microscopy .................................................................................................... 121 5.3.3 Human umbilical vein endothelial cell culture .............................................. 122 5.4 Results and Discussions ........................................................................................ 123 5.5 Conclusion............................................................................................................. 135 Chapter 6 Contributions and recommendations .............................................................. 140 6.1 Membrane chromatography .....................................Error! Bookmark not defined. x 6.2 Paper based membrane chromatography .................Error! Bookmark not defined. 6.3 Hollow fiber modification with natural hydrogel ....Error! Bookmark not defined. 6.4 Calcium alginate fiber fabrication using hollow fiber membrane Error! Bookmark not defined. xi List of Figures Figures in chapters 2, 3, 4 and 5 are as they appear in published paper, submitted manuscript, or manuscript with pending submission. Chapter 1 Figure 1.1 The comparison of mass transport and resolution in bead based column chromatography versus membrane chromatography *adopted from [1] Figure 1.2 The summary of individual research project. A) mAb polishing with PVDF membrane chromatography B) multi-proteins separation with paper based composite membrane C) calcium alginate coated on hollow fiber membrane extracapillary surface D) calcium alginate fiber fabrication with hollow fiber membrane molding technique Chapter 2 Figure 1 Schematic diagram for monoclonal antibody polishing based on hydrophobic interaction membrane chromatography Figure 2 Effect of salt concentration on the binding of hIgG1-CD4 on hydrophilized PVDF membrane (eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + different concentrations of ammonium sulfate (1) 1.2 M, (2) 1.3 M, (3) 1.4 M and, (4) 1.5 M; hIgG-CD 4 concentration: 0.1 mg/mL, injection volume: 500 μL, flow rate: 2 mL/min) xii Figure 3 Effect of salt concentration on the binding of hIgG1-CD4 “dimers” on hydrophilized PVDF membrane (eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + different concentrations of ammonium sulfate (1) 1.5 M, (2) 1.4 M, (3) 1.3 M and, (4) 1.2 M; hIgG1-CD 4 “dimers” concentration: 0.1 mg/mL, injection volume: 500 μL, flow rate: 2 mL/min) Figure 4 Effect of salt concentration on the binding of protein-A on hydrophilized PVDF membrane (eluting buffer: 20 mM sodium phosphate, pH 7; binding buffer: eluting buffer + different concentrations of ammonium sulfate (1) 1.5 M, (2) 3 M; protein-A concentration: 0.1 mg/mL; injection volume: 500 μL, flow rate: 2 mL/min) Figure 5 Effect of protein A amount on HIMC chromatograms obtained using mAb – protein-A mixtures (membrane: hydrophilized PVDF; eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + 1.37 M ammonium sulfate; chromatogram (1): 0.002 mg protein A + 0.035 mg mAb; chromatogram (2): 0.0015 mg protein A + 0.035 mg mAb; chromatogram (3): 0.001 mg protein A + 0.035 mg mAb; chromatogram (4): 0.0005 mg protein A + 0.035 mg mAb; injection volume: 500 μL, flow rate: 2 mL/min) Figure 6 Polishing of mAb by hydrophobic interaction membrane chromatography (membrane: hydrophilized PVDF; eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + 1.37 M ammonium sulfate; feed: 0.04 mg mAb monomer + 0.005 mg “dimers” + 0.0025 mg protein-A; injection volume: 500 μL, flow rate: 2 mL/min) xiii Figure 7 Size exclusion chromatography of samples obtained from the HIMC based mAb polishing experiment (1: pure mAb monomer, 2: “dimers”, 3: simulated feed, 4: flow through, 5: eluate; column: Ultrahydrogel SEC column, mobile phase: 0.25 M sodium chloride solution in water, flow rate: 0.3 mL/min) Figure 8 Coomassie blue stained SDS-PAGE gel (10%, reducing) obtained with samples from the HIMC based mAb polishing experiment (M: marker; lane 1: flow through, lane 2: eluate, lane 3: simulated feed; lane 4: pure protein-A; lane 5: “dimers”; lane 6: mAb monomer) Chapter 3 Figure 1 Effect of salt concentration on the binding of three proteins (hIgG, HSA, and Insulin) on butyl Sepharose TM HIC (eluting buffer: 20mM sodium phosphate, pH 7; binding buffer: eluting buffer+ different concentrations of ammonium sulphate – (1) 1.3 M, (2) 1.5 M; model protein concentration: hIgG 0.21 mg+ HSA 0.35 mg+ Insulin 0.14 mg in 500μL injection volume) Figure 2 Coomassie blue stained SDS-PAGE gel (10%, non-reducing) obtained with samples from the butyl Sepharose TM HIC. Multi-protein mixture (M: marker; lane 1: hIgG, lane 2: HSA, lane 3: insulin, lane 4: feed –three protein mixture, lane 5: flow through-peak a, lane 6: first elute-peak b, lane 7: second elute-peak c) Figure 3 Pressure profile in buffer change experiment carried out using filter paper based composite membranes (flow rate: 1.0 mL/min; 1.5 M ammonium sulphate in 20 xiv mM sodium phosphate buffer, 3M of sodium chloride in 20 mM sodium phosphate buffer, pH 7.0) Figure 4 Protein binding effect test using composite membrane, hIgG 0.5mg, and insulin 0.3 mg in 500 μL injection loop(flow rate: 1.0 mL/min; 0.6M and 0.8M of ammonium sulphate in 20mM sodium phosphate buffer, pH 7.0) Figure 5 Effect of salt concentration on the binding of three model proteins on PVCL composite membrane with 20 mL of gradient (eluting buffer: 20 mm sodium phosphate, pH 7.0; binding suffer: eluting buffer + 1.3 M ammonium sulphate, injected proteins concentration: IgG 0.21 mg+ HSA 0.35 mg+ Insulin 0.14 mg in 500μL injection volume, total protein loading 0.7 mg/0.25 mL of bed volume, flow rate: 1.0 mL/min) Figure 6 Size exclusion chromatography of samples obtained from the HIMC based multi-proteins separation a) pure standard samples 1: hIgG, 2: HSA, 3: Insulin, 4: feed – (hIgG+ HSA+ Insulin), b) fractions from figure 5, 1: flow through, 2: first eluting peak 3: second eluting peak; column: TSKgel G3000SWXL, mobile phase: 0.25 M sodium chloride solution in 20 mM of sodium phosphate, flow rate: 0.3 mL/min) Figure 7 Coomassie blue stained SDS-PAGE gel (10%, reducing) obtained with samples from the HIMC based multi-protein mixture (M: marker; lane 1: hIgG, lane 2: HSA, lane 3: insulin, lane 4: feed –three protein mixture, lane 5: flow through, lane 6: first elute peak, lane 7: second elute peak xv Figure 8 Effect of salt concentration on the binding of three model proteins on pVCLM composite membrane with step change 0-70-100 % (eluting buffer: 20 mm sodium phosphate, pH 7.0; binding suffer: eluting buffer + 1.3 M ammonium sulphate, injected proteins concentration: IgG 0.21 mg+ HSA 0.35 mg+ Insulin 0.14 mg in 500μL injection volume, flow rate: 1.0 mL/min) Figure 9 Size exclusion chromatography of samples obtained from the HIMC based multi-proteins separation a) pure standard samples 1: hIgG, 2: HSA, 3: Insulin, 4: feed – (hIgG+ HSA+ Insulin), b) fractions from figure 8, 1: flow through, 2: first elute peak, 3: second elute peak, column: TSKgel G3000SWXL, mobile phase: 0.25 M sodium chloride solution in 20 mM of sodium phosphate, flow rate: 0.3 mL/min) Figure 10 Coomassie blue stained SDS-PAGE gel (10%, reducing) obtained with samples from Figure 8 (M: marker; lane 1: hIgG, lane 2: HSA, lane 3: insulin, lane 4: feed –three protein mixture, lane 5: flow through, lane 6: first elute peak, lane 7: second elute peak) Chapter 4 Figure 1 The method for fabricating calcium alginate coating on ultrafiltration hollow fiber membrane as a substrate. A) cross section view of hollow fiber membrane lumen filled with water or 50 v/v% ethanol for pre-wetting step, B) after removal of water or 50 v/v% ethanol, introduce of sodium alginate solution in shell side at the mean xvi while calcium chloride solution enter the membrane lumen, C) cross-linking of alginate radially outward diffusion of calcium ions Figure 2 Experimental set-up for membrane characterization on hydraulic permeability at constant pressure, single PES hollow fiber membrane with effective length 30 cm, membrane module with effective inner volume 23.57 mL, and data acquisition (pressure and mass per time) with Logger pro. Figure 3 Experimental set-up for DMSO (300 g/L) diffusion experiment, PES ultrafiltration hollow fiber membrane with 30 cm effective length, Extracapillary space effective volume 5.89 mL, the pressure data collected and analyzed in Logger Pro (flow rate: 0.00866ml/min, pressure 101 kPa ) Figure 4 Magnified side view images of the uncoated/coated PES hollow fiber membrane. Images were obtained by light microscopy with a magnification factor of 25. Coating conditions: 1 wt% Alginate concentration, 0.1 M calcium chloride concentration in 50 v/v% ethanol, and 40 min cross-linking time Figure 5 Fabrication of calcium alginate coated hollow fiber membrane controllability test. Fabrication conditions: 1wt% alginate concentration, 0.1M of calcium chloride concentration, varied cross-linking time 0, 5, 10, 15, 20, and 40 min. Figure 6 Hollow fiber membrane coating with calcium alginate hydrogel A) Dew: calcium chloride solution pass through membrane pores B) 5 min cross-linking process C) xvii 15 min cross-linking time. Fabrication condition: sodium alginate 1 wt%, calcium chloride 0.1 M in water, PES hollow fiber membrane 150 kDa MWCO. Figure 7 TEM image obtained with the hollow fiber and composite membrane fabricated with 1 wt% sodium alginate and 0.1 M calcium chloride on PES ultrafiltration membrane, with a cross-linking time of 30 min. (A) control sample: uncoated membrane,25000 X magnification (B) 1 wt % of sodium alginate and 0.1 M calcium alginate in water, 5000 X magnification (C) bigger magnification of sample B, 20000X (D) 0.1M of calcium chloride solution in 50 v/v EtOH %, 20000 X magnification Figure 8 Calcium alginate polymerization study with different calcium chloride solvents (A: alginate + water based calcium chloride, B: alginate + 50v/v% ethanol based calcium chloride, C: alginate + 100 % ethanol, D: ethanol + alginate, E: sample D vertical pose), 1 wt% sodium alginate of 1mL, 0.1 M calcium chloride 300 μL Figure 9 Fabrication of calcium alginate coated on PES hollow fiber membrane (Alginate concentration 1wt%, calcium chloride concentration 0.1M and 1 min crosslinking time) optical microscope 25 times. Figure 10 Water contact angle analysis with PES hollow fiber membrane (left), and calcium alginate coated membrane (right). Picture obtained by digital camera. Figure 11 Measurement of water flux and hydraulic permeability of uncoated/coated PES hollow fiber membrane, operating pressure at 109.45 kPa (uncoated membrane) and 110.56 kPa (coated membrane) with constant. xviii Figure 12 Effect of coating hollow fiber membrane on DMSO mass transport with Fig 3 experimental set up. Chapter 5 Figure 1 The method for fabricating calcium alginate hollow fibers using porous hollow fiber membrane as mold (A) cross sectional view of hollow fiber membrane with pores filled with calcium chloride solution, (B) sodium alginate solution being filled inside the lumen of the hollow fiber membrane, (C) cross-linking of alginate by radially inward diffusion of calcium ions, (D) removal of un-cross-linked alginate core with calcium chloride solution, and (E) expulsion of calcium alginate hollow fiber from mold. Figure 2 Sequence of events in calcium alginate hollow fiber molding process (A) sweating of calcium chloride on the outer wall of the hollow fiber membrane indicating that the pores are filled, (B) removal of un-cross-linked sodium alginate core from the mold using calcium chloride solution, (C) expulsion of calcium alginate hollow fiber from mold using water, (D) alginate hollow fiber after recovery from mold. Figure 3 Alginate fibers fabricated using hollow fiber membrane based molding technique: (A) Fibers made with 2.5% sodium alginate, 0.1 M calcium chloride solution as cross-linker using (from the top) Accurel Q3/2, Tetronic and Accurel PP S6/2 hollow fiber membrane molds respectively. The cross-linking times were 10, 30 and 60 seconds respectively for the small, medium and large diameter hollow fibers. (B)Solid and (C) hollow fibers fabricated with 5% sodium alginate, 1 M calcium chloride solution as xix cross-linker, using Accurel PP S6/2 membrane as mold. The cross-linking times were 600 seconds and 60 seconds respectively. Figure 4 Magnified cross-sectional and side view images of the alginate hollow fiber shown in Figure 3 C. Images were obtained with the fiber immersed in water by light microscopy with a magnification factor of 25. Figure 5 Confocal laser scanning (CLS) microscopy images of the FITC-dextran - alginate composite hollow fiber, (A) z-stack images obtained along the axis at sequential depths of 200μm, (B) five times magnification, and (C) twenty-time magnification Figure 6 Scanning electron microscopy (SEM) images of calcium alginate hollow fiber (400X magnification) fabricated with 2.5 % sodium alginate and 1 M calcium chloride, using Accurel PP S6/2 membrane as mold, with a cross-linking time of 60 seconds. Figure 7 TEM image obtained with the hollow fiber described in Figure 6 at (A) 5000X magnification, and (B) 75000X magnification Figure 8 Effect of sodium alginate concentration on dimension of alginate fiber (mold: Accurel PP S6/2, calcium chloride concentration: 1 M; cross-linking time: 60 seconds) xx Figure 9 Effect of calcium chloride concentration on dimension of alginate fiber (mold: Accurel PP S6/2, sodium alginate concentration: 5%, cross-linking time: 60 seconds) Figure 10 Effect of cross-linking time on dimensions of alginate fiber (mold: Accurel PP S6/2, sodium alginate concentration: 5%, calcium chloride concentration: 1 M) Figure11 Light microscopy image of HUVEC embedded alginate hollow fiber (image obtained one day after fabrication, magnification: 100 X, sodium alginate concentration: 1%, mold: Tetronic, calcium chloride concentration: 0.1 M, cross-linking time: 30 seconds) List of Tables Tables in chapter 2, 3, 4 and 5 are as they appear in published paper, submitted manuscript, or manuscript with pending submission. Chapter 1 Table 1. Membrane specifications in this dissertation Chapter 2 Table 1. Different combinations of protein-A and mAb used in HIMC experiments represented in Fig. 5. xxi Chapter 4 Table 1. PES hollow fiber membrane coated with calcium alginate; 0.1 M calcium chloride solution was prepared in 50 v/v% ethanol, 1 wt % sodium alginate, in different cross-linking time Nomenclature E1 First eluted peak E2 Second eluted peak FT Flow through HSA Human serum albumin HIC hydrophobic interaction chromatography HIMC hydrophobic interaction membrane chromatography HPLC High pressure liquid chromatography kDa kilo Dalton LCST Lower critical solution temperature MWCO Molecular weight cut-off PEG Poly(ethylene glycol) xxii PVCL Poly(N-vinylcaprolactam) PVDF Polyvinylidene fluoride SDS-PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis SEC Size exclusion chromatography T Temperature (°C) P Pressure (kPa) xxiii Preface This Ph.D. thesis is organized in a sandwich style based on the following published, submitted and prepared articles: 1. S.M. Yoo, R. Ghosh, Simultaneous removal of leached protein-A and aggregates from monoclonal antibody using hydrophobic interaction membrane chromatography, J.Membr. Sci, 390-391 (2012) 263-269. This article was made into Chapter 2. 2. S.M. Yoo, R. S.H. Park, R. Ghosh, High-resolution hydrophobic interaction chromatographic separation of multi-protein mixture using paper-based composite membranes. Manuscript is in preparation. This article was made into Chapter 3. 3. S.M. Yoo, R. Ghosh, Hollow fiber membrane surface coating with natural hydrogel for bioreactor, Manuscript in preparation. This article was made into Chapter 4. 4. S.M. Yoo, R. Ghosh, Fabrication of alginate fibers using a microporous membrane based molding technique, in submitted in Biochem. Eng. J. Manuscript # BEJ-D-1400034R1. This article was made into Chapter 5. All the other articles were prepared by Seung Mi Yoo. Dr. Ghosh provided guidance in research direction and project planning, and also helped in manuscript preparation. Ryan Sung Ho Park conducted initial experiments, membrane characterization and protein binding, as a project student for the project described in Chapter 3. The work reported in this dissertation was undertaken from January 2013 to April 2014. xxiv Chapter 1. Introduction of membranes with hydrogels 1.1 Membrane technology Membrane technology has experienced tremendous application in chemical and biopharmaceutical engineering industries. The first significant industrial application of membrane technology took place in 1960s, and ever since then it has contributed significantly to improvement in quality of life [1]. There are various definitions for the term ‘membrane’ depending on the application it is being used, for example simply filtering particles or selectively transporting one or more substances. For a biochemical engineer who has studied downstream processing, a membrane is a physical separation barrier with porous structure which requires either pressure or another driving force to selectively separate materials. A membrane is generally defined as “a thin barrier or film through which solvent and solutes are selectively transported” [2]. In the biopharmaceutical industry membranes have been used for protein concentration, microorganism and virus removal, buffer exchange, and cell recovery [3]. A much simpler mechanism is utilized in the water treatment industry, where dispersed particles are removed by size exclusion or sieving [4]. There are many membrane manufactures involved in the production of different types of membrane elements and configurations that are used for various applications. The major three international membrane companies whose products have been used in my research are Merck Millipore, and Membrana 1 GmbH. Microfiltration and ultrafiltration membranes from these companies that have been used in my projects are made of organic polymers such as hydrophilized polyvinylidene fluoride (PVDF) (Chapter 2), cellulose (Chapter 3), polyethersulfone (PES) (Chapter 4 and 5), polypropylene (PP) (Chapter 5), polysulfone (PS), cellulose acetate (CA), and regenerated cellulose (see Table 1). Membranes are generally available in three geometries: flat sheet (Chapter 2 and 3), hollow fiber (Chapter 4 and 5), and tubular (not discussed in this thesis). The advantage of a flat sheet membrane is that it can be utilized for various types of operations in different module configurations. For example, a flat sheet membrane could be cut into a desired shape (square, circular, or rectangular) and size to fit a module for dead-end, cross flow, or co/counter current flow separation, whereas hollow fiber and tubular membrane are available pre-assembled within membrane modules. Membranes are also categorized based on their pore sizes into micro-, ultra-, nanofiltrations, and reverse osmosis. Micro- and ultra- filtration are most extensively used for waste water treatment and for biopharmaceutical production. Nanofiltration and reverse osmosis are used in clinical setting such as viral removal or plasma-derived product separation [43] and drinking water purification units. Commercial microfiltration membranes commonly have 0.45, 0.2, 0.1 μm pore sizes which are able to reject microorganism from feed solutions. Using this rejection characteristic, buffers and other solutions can be clarified. Ultrafiltration is mainly employed for protein concentration 2 and buffer exchanges prior to analysis. Table 1 shows the specifications of the membrane that were used in the different projects discussed in this thesis. Table 1 Membrane specifications in this dissertation Flat sheet Hollow fiber Material Catalogue information Modification method/base material Thickness Manufacture Morphology Hydrophilized PDVF GVWP 14250 Grafted 125 µm Millipore asymmetric Composite membrane Fabricated Coating PVCL on filter paper 200 µm Fabricated symmetric Polypropylene Accurel PP S6/2 Polypropylene Membrana Isotropic Accurel PP Q3/2 Polypropylene Membrana Isotropic Tetronic Hydrophilized PES 1.8 mm i.d./2 mm o.d. 0.6 mm i.d./1mm o.d. 0.8 mm i.d./ 1.3 mm o.d. Hydranautics Anisotropic Polyethersulfone The membrane industry has been producing high quality membrane for more than 50 years. However, while some product lines have remained the same, the applications and operating conditions required better mechanical/ chemical durability, high flux, less fouling, and easy to operation. To solve these problems, many researchers and membrane developers are still seeking for new types of membrane such as hydrogel based composite membrane, which will be discussed in considerable detail in this thesis. 1.2 Membrane chromatography Various bioseparation techniques are used in the downstream processes of the biopharmaceutical industry. Column chromatography has been used extensively in 3 production lines for the separation and purification of biological samples with high resolution. For example, various types of column chromatography are used in the capture (affinity chromatography), purification (hydrophobic interaction chromatography), and polishing (ion exchange chromatography) steps of antibody purification [5]. However, because the separation relies mainly on the diffusion of samples through pores in a soft gel matrix, the procedure requires long contact times with large volumes of mobile phase. In addition, when this soft material is placed in a column, the drop in pressure across the column results in the deformation of the beads. These disadvantages limit the scalability of this technique. Therefore, studies have focused on advancing the technique by improving the diversity of the support matrix, narrow pore distribution, and ligand chemistry. Although new modifications in the method, such as combining different kinds of column chromatography for more complex protein separations have been investigated, the operating conditions have a narrow window, and possible target proteins are very limited. A simulated moving bed (SMB) technique for multi-protein separations was developed in the early 2000s [6]. However, this technique has limited applications. The operating condition requires the ratio of the individual protein or amino acid residence time versus the zone flow rate to be calculated and estimated from many parameters. Moreover, it requires tedious optimization procedures for particular target proteins. Membrane chromatography provides an attractive and efficient mass transport process, driven predominantly by the principle of convection flow, as illustrated in Figure 1.1. The membrane adsorbers that are used in this technique possess high-throughput abilities. 4 These membrane stacks create void channels for the mobile phase to pass, allowing selective separation of biological substances. Membrane chromatography incorporates aspects of membrane filtration (high-throughput) and packed-bed column chromatography (high-resolution) [7, 8]. The combination of membrane and column chromatography was developed to achieve better separation with smaller footprints, shorter process times, less buffer usage, and more flexibility in terms of configuration and operation. Furthermore, the limitations associated with conventional column chromatography, such as sudden drops in pressures, may be minimized. The relatively rigid microporous structure of the membrane allows for a high flux and minimal batch-tobatch variations [2]. Figure 1.1 Comparison of the mass transport process and resolution in bead-based column versus membrane chromatography. *adopted from [9]. 5 Membrane chromatography operates on the same principle as column chromatography. The affinity moiety is immobilized on the membrane substrate and serves as the stationary phase. The mobile phase (protein mixture or biological sample) is then passed over the stationary phase. The sample interacts with the stationary phase and binds to the membrane surface, while the impurities remain in the flow-through. The sample is then eluted from the surface of the membrane by changing the operating conditions such that the interaction between the sample and the affinity moiety is broken. These membranes may be used as alternatives to protein-A/G affinity column chromatography for the purification of antibodies [10, 11]. In addition, ion exchange membrane chromatography may be used to remove impurities such as host cell protein (HCP), DNA, and viruses from biological samples [12, 13]. A number of recent studies have demonstrated the use of hydrophobic interaction membrane chromatography (HIMC) [14–17] for the purification of monoclonal antibodies (mAbs) [18–26]. Ghosh (2001) used a polyvinylidene fluoride (PVDF) membrane to separate CAMPATH-1G monoclonal antibodies and bovine serum albumin (BSA) using hydrophobic interactions [18]. Similarly, human plasma proteins, human immunoglobulin G (hIgG) and human serum albumin (has) could be separated using a PVDF membrane (pore size, 1 μm) [27]. A paper-polyethylene glycol (PEG) interpenetrating polymer network-based composite membrane was also developed as an inexpensive alternative to commercial membranes [22]. The PEG polymer undergoes a phase transition when treated with high concentrations an anti-chaotropic salt; this 6 characteristic feature of the polymer was adopted to separate and purify mAb on the basis of its hydrophobic interaction with the PEG polymer. Membranes have not only been examined for their adsorption capacity and resolution but also for their binding mechanisms, which were investigated using rabbi IgG and proteinA conjugates [21]. The binding sites of these conjugates on the membrane surface were identified by digesting them with papain and pepsin. Because protein-A recognizes the Fc domain of rabbit IgG, the Fab domain and the hinge region of IgG are available for interaction with the membrane. This binding mechanism was analyzed using a confocal laser-scanning microscope. In their studies, Huang et al. (2009) coated a PVDF membrane with an environmentally responsive polymer (poly-N-vinylcaprolactam [PVCL]) for use in HIMC [24]. Pereira et al. (2010) demonstrated that a modified Sartobind® epoxy membrane supported with a linear alkyl chain ligand could be used for plasmid DNA purification [23]. The alkyl ligand layer increased the adsorption of RNA on the membrane, as expected. Other studies investigated the use of composite membranes such as paper-based membranes for the purification of mAb [25]. Environmentally responsive PVCL was grafted on regenerated cellulose membranes using the atom-transfer radical-polymerization (ATRP) technique; the binding capacity per column volume for IgG and HSA were then analyzed [25, 26]. Two parameters of composite membranes may be manipulated by environment stimuli: the range of binding capacity and the permeability. 7 Membrane chromatography is widely used commercially for the purification and fractionation of biological samples in the laboratory as well as the pilot-scale. Mustang® ion exchange membrane (Pall Life Sciences), Sartobind® Phenyl, and Sartobind® S/Q (Sartorius AG) are available commercially. Mustang® Q or S (or Sartobind® S or Q) are based on the principles of ion exchange chromatography. Similarly, Sartorius also manufactures an HIMC membrane composed of regenerated cellulose with covalently attached phenyl ligands, called Sartobind® Phenyl [28]. This commercial product is prestacked within the cartridge for convenience; however, certain aspects such as fluid distribution within the membrane module and scale-up with respect to bed volume need to be improved. Moreover, commercially available products are expensive to use, whereas conventional chromatography columns may be regenerated and reused approximately 100 times, depending on the chemistry involved. Two operating modes are used in membrane chromatography: the flow-through mode and the bind-elute mode. The flow-through mode has been embraced to remove trace amounts of unwanted substances from mAb purifications [12, 20]. This mode has the advantage of simple operating conditions, where the desired products are collected in the eluate and trace amounts of impurities are entrapped within the membrane matrix. This can be achieved without changing the mobile fluid conditions and does not require a membrane regeneration step. The flow-through mode is excellent for use as the final polishing step for biopharmaceuticals; however, the selectivity of the membrane has to be high enough to separate the product from the impurities and the membrane quality and operating 8 conditions have to be precise. In mAb production, a protein-A affinity column is the main capturing tool used to separate mAb from HCP, DNA, or viruses. The protein-A ligand binds strongly to the Fc domain of mAbs; therefore, the capture step is the most crucial step in mAb production. Despite the benefits associated with the use of protein-A column process, the low pH conditions required for dissociation cause the mAbs to aggregate and leach protein-A ligand. Therefore, an ion exchange membrane chromatography technique was developed for the large-scale production of mAbs [12]. At neutral pH, the impurities and mAbs have different charges (negative and positive, respectively). Therefore, anion exchange membrane chromatography may be used to purify mAbs. Knudsen et al. (2001) reported that cation and anion exchange membranes may be utilized for the removal of potential contaminants and impurities. However, because cation exchange membrane chromatography has to be operated under two operating steps (bind and elute), the operating cost increases compared to anion exchange membrane chromatography (available commercially as Sartobind® Q) [12]. The economic aspect of using membrane chromatography was reviewed by Zhou and Tressel in 2006 [29]. Buffer usage may be lowered by as much as 95% with membrane chromatography compared to packed-bed anion exchange chromatography. The disposability of membrane columns eliminates the packing, washing, and regeneration steps of column chromatography, thereby reducing processing time and labor costs. A long-term cost calculation analysis of membrane versus resin-based column chromatography revealed that membrane chromatography is cheaper than column chromatography. 9 The bind-elute operating mode has been actively investigated for use in both academia and industry. This operating mode requires the target substance to be bound on the membrane, followed by elution in one or multiple steps or, alternatively, in a gradient. However, membranes (in the absence of hydrogel-modifying treatments) have a lower binding capacity than resin-based columns; therefore, improvements in binding capacity were required. The most common application of the bind-elute mode is in affinity membrane chromatography, which has replaced protein-A/G column chromatography for the purification of mAb [10, 11]. The bind-elute mode has also been applied in HIMC for the separation of proteins [18,19,21,22,25]. Recently, hydrogel coated/grafted membranes are being actively investigated because they provide improved binding capacities. The development of a hydrogel composite membrane is discussed in the next section. 1.3 Hydrogel based membrane Synthetic membranes are used widely in the environmental and biopharmaceutical industries. Membrane adsorbers, which are traditionally organic-based materials, have been developed using the emerging hydrogel technology for enhanced performance and functionality. Two types of hydrogels have been studied for use in hydrophobic interaction, ion exchange, and affinity membrane chromatography. One is the synthetic, polymer-based hydrogel, which may be composed of PEG, poly (N-isopropylacrylamide) (PNIPAM), PVCL, polyester, poly lactic acid (PLA), poly lactic-co-glycolic acid (PLGA), and polycarbonate. The other type comprises naturally derived hydrogels, such 10 as those based on dextran, alginate, collagen, fibrin, and gelatin [30]. Hydrogels are cross-linked water-soluble polymers with hydrophilic pendent groups such as amides and carboxylic acids in their structure. The combination of the hydrophobic membrane substrate and the hydrophilic hydrogel resulted in the development of environmentally responsive membranes, also termed “smart-membranes.” Various environmentally responsive membranes have been developed by many researchers [14, 21, 24, 26, 31–33]. Such membranes change their physical structure, charge, or affinity in response to changes in pH [34], temperature [33], and salt concentrations [32]. By manipulating the tunable characteristics of an environmentally responsive membrane, complex or multi-component separations may be achieved easily. PNIPAM has been actively researched for cross-linking or interpenetrating membrane substrates to make a three dimensional formation [33, 35, 36]. This hydrogel network can change its physical and chemical properties upon receiving the various environmental stimuli mentioned above. Rao et al. (2002) constructed a hybrid membrane based on silica substrate with PNIPAM encapsulated within it using a sol-gel process. The permeability of the membrane changed with temperature. However, after PNIPAM toxicity became problematic in biological applications, PVCL and PEG were investigated as alternatives to PNIPAM [37, 38]. PVCL is also a thermo-responsive polymer with a low critical solution temperature (LCST). It is relatively less toxic with enhanced physical strength. It was studied extensively for the fabrication of composite membranes. However, in terms of practical applications, controlling salt concentration is much 11 simpler than controlling the temperature of the mobile phase and its environment to achieve switchable hydrophobic interactions between proteins and polymers [24]. Therefore, an inexpensive composite membrane with cellulose filter paper was developed for protein separations. Yu et al. (2008) demonstrated the use of PEG-coated cellulose paper for the separation of mAbs from Chinese hamster ovary (CHO) cell culture [22]. Mah and Ghosh (2010) demonstrated the feasibility of a composite membrane composed of PVCL on Whatman filter paper for the purification of IgG from simulated CHO cell culture. In addition to coating, hydrogels may be grafted on membrane substrates for better membrane resolution [39–42]. Although the grafting method is more complicated, involving the use of various chemicals (acids and bases) and long periods of pretreatment before polymerization, the ligand length and density may be tailored for exact applications [15]. Himstedt et al. (2013) grew PVCL polymer chains on a regenerated cellulose membrane. BSA and IgG, which are often used as model proteins, were employed to test the adsorption as well as the binding capacity of the membrane [26]. 1.4 Thesis outline This thesis consists of six chapters. Chapter 1 is an introduction to membrane technology, membrane modifications, and membrane chromatography which will also touch on all the research projects in chapter 2, 3, 4, 5. Four chapters could be categorized into two key compartments: flat membrane and hollow fiber membrane configurations. All of the 12 projects involved the combination of both hydrogels and membranes. Chapter 2 and chapter 3 focus on membrane chromatography for various protein separation applications. Chapter 4 and 5 are studied on membrane modification with natural hydrogel for tissue engineering applications. The summary of each project is shown in Figure 1.2. Figure 1.2 The summary of individual research project. A) mAb polishing with PVDF membrane chromatography B) multi-proteins separation with paper based composite membrane C) calcium alginate coated on hollow fiber membrane extracapillary surface D) calcium alginate fiber fabrication with hollow fiber membrane molding technique Chapter 2 describes the purification of a monoclonal antibody from its aggregates and leached protein-A using membrane chromatography. In this project, the membrane chromatography consisted of a hydrophilized PVDF membrane which is commercially available in the market as an ultrafiltration membrane. A PEG hydrogel, which had environment responsiveness to salt present in solution, was grafted into the pores of the hydrophilized PVDF membrane substrate [29]. This hydrogel, which was based on an 13 LCST polymer, had a phase transition controlled by salt concentration. With stimuli responsiveness, apparent protein and grafted polymer hydrophobic interaction degree changed. Manipulating the salt concentration made the pure monoclonal antibody separate from impurities in flow-through mode. This work was published in Journal of Membrane Science. Chapter 3 presents the fractionation of plasma proteins, hIgG, and HSA, and also a popular hormone, insulin, using a paper based composite membrane. A composite membrane was fabricated using poly (N-vinylcaprolactam) polymer coated on Whatman cellulose filter paper. Membrane chromatography was tested for environment responsive characteristics under different ionic strength, and the transmembrane pressure was a good indication on the hydrogel states. The collapsed and swollen states of hydrogel induced a hydrophobic difference on membrane matrix. By manipulating this characteristic, protein had experienced different interaction with the membrane. This work is ready for journal submission. Chapter 4 investigated a hollow fiber membrane extracapillary surface coated with calcium alginate. The polymerization method is novel and the thickness of the coating layer is highly controllable using a gentle and precise procedure. The cross-linkers are passively diffused through the membrane pores and come into contact with the crosslinking alginate in extracapillary space. This composite membrane was studied for membrane performance on hydraulic permeability and solute mass transport rate. The hydrophilic characteristic was tested using water contact angle. The TEM analysis was 14 obtained for calcium alginate morphologies. This work also was written in manuscript for journal submission. Chapter 5 shows new application of micro- or ultra-filtration hollow fiber membrane. The membrane pores were utilized for storage of cross-linking reagent, calcium ion, when monomer (sodium alginate) solution contacted the polymerization process occurred. A hollow fiber membrane is used as the mold for fabricating these calcium alginate fibers. This process was used for membranes of varying diameters, sizes, and geometries. This work has been submitted in Biochemical Engineering Journal. As a whole, the research reported in this thesis could lead to four journal publications. One is published (Yoo and Ghosh 2012, Journal of Membrane Science), one is submitted (in Biochemical Engineering Journal, BEJ-D-14-00034), and two more papers are ready submission to journals. 15 1.5 References [1] R.Baker, Overview of membrane science and technology, in: Membr. Technol. Appl., 2nd ed., Wiley, England, 2004: pp1-14. [2] R. Ghosh, Membranes, in: Protein bioseparation using ultrafiltration, Imperial College Press, London, UK, 2003: pp 19. [3] P. Gagnon, Purification tools for monoclonal antibodies by mixed-mode chromatography, in: Process scale purification of antibodies, U. Gottschalk, ed., John Wiley & Sons, New York, 2009 pp 125-144. [4] J.C. Schrotter, B. Bozkaya-Schrotter, Current and emerging membrane processes for water treatment, in: Membrane for water treatment, K.V. Penemann, S.P. Nunes, John Wiley & Sons, Weinheim, 2010 pp 53-91. [5] B. Kelley, G. Blank, A. Lee, Downstream processing go monoclonal antibodies: Current Practices and future opportunities. In: Process scale purification of antibodies. John Wiley & Sons, Hoboken, NJ, 2009 pp1-23. [6] D. Wu, Y. Xie, Z. Ma, N.L. Wang, Design of simulated moving bed chromatography for amino acid separations, 5885 (1998) 4023-4035. [7] J. Thommes, M.R. Kula, Membrane chromatography-an integrative concept in the downstream processing of proteins, Biotechnol. Prog. 11 (1995) 359-367. [8] J.A. Gerstner, R. Hamilton, S.M. Cramer, Membrane chromatographic protein separations systems for high-throughout, 596 (1992) 173-180. [9] R. Ghosh, Protein separation using membrane chromatography: opportunities and challenges, J. Chromatogr. A. 952 (2002) 13-27. [10] H. Zou, Q. Luo, D. Zhou, Affinity membrane chromatography for the analysis and purification of proteins, J. Biochem. Biophys. Methods. 49 (2001) 199-240. [11] L.R. Castilho, F.B. Anspach. W.D. Deckwer, Comparison of affinity membranes for the purification of immunoglobulins, J. Membr. Sci. 207 (2002) 253-264. [12] H.L. Knudsen, R.L. Fahrner, Y. Xu, L.A. Norling, G.S. Blank, Membrane ionexchange chromatography for process-scale antibody purification, 907 (2001) 145154. 16 [13] M. Phillips, J. Cormier, J. Ferrence, C. Dowd, R. Kiss, H. Lutz, J. Carter, Performance of a membrane adsorber for trace impurity removal in biotechnology manufacturing, J. Chromatogr. A. 1078 (2005) 74-82. [14] J. Queiroz, C.T. Tomaz, J.M. Cabral, Hydrophobic interaction chromatography of proteins, J. Biotechnol. 87 (2001) 143-159. [15] A. Ladiwala, F. Xia, Q. Luo, C.M. Breneman, S.M. Cramer, Investigation of protein retention and selectivity in HIC systems using quantitative structure retention relationship models, Biotechnol. Bioeng. 93 (2006) 836–50. [16] D.K. Roper, E.N. Lightfoot, Separation of biomolecules using adsorptive membranes, J. Chromatogr. A. 702 (1995) 3–26. [17] R. van Reis, A. Zydney, Membrane separations in biotechnology, Curr. Opin. Biotechol. 12 (2001) 208-211. [18] R. Ghosh, Separation of proteins using hydrophobic interaction membrane chromatography, J. Chromatogr. A. 923 (2001) 59–64. [19] R. Ghosh, L. Wang, Purification of humanized monoclonal antibody by hydrophobic interaction membrane chromatography, J. Chromatogr. A. 1107 (2006) 104–9. [20] S.M. Yoo, R. Ghosh, Simultaneous removal of leached protein-A and aggregates from monoclonal antibody using hydrophobic interaction membrane chromatography, J. Membr. Sci. 390-391 (2012) 263–269. [21] X. Sun, D. Yu, R. Ghosh, Study of hydrophobic interaction based binding of immunoglobulin G on synthetic membranes, J. Membr. Sci. 344 (2009) 165–171. [22] D. Yu, X. Chen, R. Pelton, R. Ghosh, Paper-PEG-based membranes for hydrophobic interaction chromatography: purification of monoclonal antibody, Biotechnol. Bioeng. 99 (2008) 1434–42. [23] L.R. Pereira, D.M.F. Prazeres, M. Mateus, Hydrophobic interaction membrane chromatography for plasmid DNA purification: Design and optimization, J. Sep. Sci. 33 (2010) 1175–84. [24] R. Huang, K.Z. Mah, M. Malta, L.K. Kostanski, C.D.M. Filipe, R. Ghosh, Chromatographic separation of proteins using hydrophobic membrane shielded with an environment-responsive hydrogel, J. Membr. Sci. 345 (2009) 177–182. 17 [25] K.Z. Mah, R. Ghosh, Paper-based composite lyotropic salt-responsive membranes for chromatographic separation of proteins, J. Membr. Sci. 360 (2010) 149–154. [26] H.H. Himstedt, X. Qian, J.R. Weaver, S.R. Wickramasinghe, Responsive membranes for hydrophobic interaction chromatography, J. Memb. Sci. 447 (2013) 335–344. [27] R. Ghosh, Fractionation of human plasma proteins by hydrophobic interaction membrane chromatography, J. Memb. Sci. 260 (2005) 112–118. [28] N. Fraud, R. Faber, C. Kiss, Novel membrane adsorber for hydrophobic interaction chromatography, in: Proceedings of the 236th ACS National Meeting, Philadelphia, PA, USA, 2008. [29] J.X. Zhou, T. Tressel, Basic concepts in Q membrane chromatography for largescale antibody production, Biotechnol. Prog. (2006) 341-349. [30] K.Y. Lee, D.J. Mooney, Hydrogels for Tissue Engineering, Chem. Rev. 101 (2001) 1869–1880. [31] R. Huang, L.K. Kostanski, C.D.M. Filipe, R. Ghosh, Environment-responsive hydrogel-based ultrafiltration membranes for protein bioseparation, J. Memb. Sci. 336 (2009) 42–49. [32] L.K. Kostanski, R. Huang, R. Ghosh, C.D.M. Filipe, Biocompatible poly(Nvinyllactam)-based materials with environmentally-responsive permeability, J. Biomater. Sci. Polym. Ed. 19 (2008) 275–90. [33] D, Crespy, M. Rossi, Temperature-responsive polymers with LCST in the physiological range and their applications in textiles, Polym. Int. 1468 (2007) 1461-1468. [34] N.M. Ranjha, J. Mudassir, Swelling and aspirin release study: cross-linked pHsensitive vinyl acetate-co-acrylic acid (VAC-co-AA) hydrogels, Drug Dev. Ind. Pharm. 34 (2008) 512–521. [35] R. Chennamsetty, I. Escobar, X. Xu, Characterization of commercial water treatment membranes modified via ion beam irradiation, Desalination. 188 (2006) 203–212. [36] G.V.R. Rao, M.E. Krug, S. Balamurugan, H. Xu, Q. Xu, G.P. Lo, Synthesis and Characterization of Silica - Poly ( N -isopropylacrylamide ) Hybrid Membranes : Switchable Molecular Filters, Chem. Mater. (2002) 5075–5080. 18 [37] H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu, J. Hirvonen, Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam)., Biomaterials. 26 (2005) 3055–3064. [38] J. Kopecek, Hydrogel biomaterials: a smart future?, Biomaterials. 28 (2007) 5185– 5192. [39] Y.J. Shen, G.X. Wu, Y.B. Fan, H. Zhong, L.L. Wu, S.L. Zhang, X.H. Zhao, W.J. Zhang, Performances of biological aerated filter employing hollow fiber membrane segments of surface-improved poly (sulfone) as biofilm carriers, J. Environ, Sci. (China) 19 (2007) 811-817. [40] K. Asfardjani, Y. Secui, Y. Aurelle, N. Abidine, Effect of plasma treatments on wettability of polysulfone and polyetherimide, 43 (1991) 271–281. [41] D.S. Wavhal, E.R. Fisher, Membrane surface modification by plasma-induced polymerization of acrylamide for improved surface properties and reduced protein fouling, Langmuir, 19 (2003) 79-85. [42] J. Chen, J. Li, C. Chen, Surface modification of polyvinylidene fluoride (PVDF) membrane by low-temperature plasma with grafting styrene, Plasma Sci. Technol. 11 (2009) 42-47. [43] T. Burnouf, M. Radosevich, Nanofiltration of plasma-derived biopharmaceutical products, Haemophilia 9 (2003) 24-37 19 Chapter 2 Simultaneous removal of leached protein-A and aggregates from monoclonal antibody using hydrophobic interaction membrane chromatography1 1 This chapter is version of the paper published in Journal of Membrane Science, 390-391: 263-269 (2012) by Seung Mi Yoo, Raja Ghosh. 20 2.1 Abstract Protein-A affinity chromatography is the standard capture technique used for purification of monoclonal antibodies (mAbs). A major problem with this technique is its inability to remove monoclonal antibody aggregates from the monomeric form of the antibody. Moreover, the elution of mAbs from a protein-A column is carried out using acidic conditions which in turn could accelerate the formation of antibody aggregates and causes leaching of protein-A from its supporting media. These impurities have to be removed using appropriate separation methods before the mAb can be used for therapeutic applications. There is currently a limited repertoire of polishing techniques which simultaneously remove both mAb aggregates and protein-A. In this paper, we describe a polishing method based on hydrophobic interaction membrane chromatography for simultaneously removing these impurities. The flow-through mode was used whereby monomeric mAb flowed through a membrane stack while impurities were retained by reversible adsorption. These impurities were subsequently eluted by lowering the salt concentration and the membrane stack was thus regenerated. The operating conditions for this method were systematically optimized using pure mAb, aggregates, protein-A, and mAb/protein-A complexes. The mechanisms by which aggregates and leached protein-A were simultaneously removed are hypothesized. 21 2.2 Introduction Monoclonal antibodies or mAbs were first produced in the 1970s [1]. These are now extensively used in biomedical research, and for a range of therapeutic and diagnostic applications [2]. MAbs are usually purified using either protein-A or protein-G based affinity column chromatography as the principal capture technique [3, 4]. Protein-A, which was originally isolated from a bacteria recognizes and binds the Fc domain of the immunoglobulin G or IgG class mAbs at neutral pH [3]. The use of protein-A affinity chromatography results in highly selective mAb purification and there is typically around 90 - 95% removal of impurities such as media proteins, host cell protein, nucleic acids (mainly DNA) and endotoxin [5]. When the mAb containing feed solution is passed through a protein-A column, the impurities are removed in the flow through while the mAb is retained within the column. The column-bound mAb is then eluted by lowering the pH, this acidic condition being suitable for inactivating viral contaminants. The mAb yield in protein-A affinity chromatography tends to be quite high. Despite these obvious advantages, the use of protein-A affinity chromatography for mAb purification throws up some challenges that need to be addressed. Firstly, protein-A does not discriminate between the mAb monomer and its aggregated forms as the Fc region is not affected by the aggregation process [6, 7]. Consequently, the mAb aggregates that are formed during cell culture and the earlier downstream processing steps such as centrifugation or microfiltration are not removed in the flow through and are found to coelute from the protein-A column along with the mAb monomer. Secondly, the lowering 22 of pH during elution accelerates mAb aggregation [8]. Aggregated forms of mAb have lower biological activity and can in some cases be toxic or immunogenic [6, 9]. Thirdly, protein-A is known to leach out in small amounts from its support matrix, more so under acidic conditions [10]. This leachate and its fragments are immunotoxic and their presence in therapeutic products is highly undesirable [11-13]. For this reason, polishing of protein-A affinity chromatography purified mAb become absolutely essential, i.e. for the removal of product related impurities such as mAb aggregates and process related impurities such as leached protein-A and its fragments. Aggregated forms of mAbs are commonly removed by size exclusion chromatography (or SEC) [14, 6]. The dimer and higher aggregates being larger are excluded from the pores present in the chromatographic media and consequently move faster through the column than the mAb monomer. The use of SEC for both preparative and analytical scale separation of mAb aggregates has been reported [14, 6]. Some of the problems of using SEC for preparative separation are low speed, poor scalability and limited resolution. Researchers have tried to address these by combining SEC with other technique [15, 16]. Techniques such as field flow fractionation (or FFF) [17] and hydroxyapatite (or HA) based column chromatography have also been used for removing aggregated forms of monoclonal antibody [18, 19]. The use of ion exchange chromatography in the presence of polyelectrolyte additives such as polyvinylsulfonic acid, polyacrylic acid and polystyrenesulfonic acid for removing mAb aggregates has been reported [20]. Wang et al. [7] used hydrophobic interaction membrane chromatography (or HIMC) for separating 23 mAb monomer from its dimer based on their hydrophobicity difference. In a subsequent paper [21] the feasibility of fractionating different aggregated forms such as dimer, trimer, tetramer and pentamer from each other using the same technique was also demonstrated. There are several other more recent reports on the use of hydrophobic interaction chromatography or hydrophobic charge induction chromatography employing both columns and membranes for the removal of mAb aggregates [22-26]. The use of the hydrophobic charged induction mode led to improvement in binding capacity, step yield, and impurity removal rate [24]. In addition, less amount of salt was required when compared with standard hydrophobic interaction chromatography. The leaching of protein-A which co-elutes along with the mAb was systematically studied by Bloom et al. [27]. They also examined the relationship between protein-A contamination level and column usage. Protein-A is typically leached in small amounts and its detection can in some cases be challenging. Steindl et al. [28] used sandwich-type enzyme linked immusorbent assay to measure extremely low concentrations of leached protein-A. However in most cases, standard techniques such as sodium dodecyl sulphate polyacrylamide gel electrophoresis (or SDS-PAGE) are considered sensitive enough to detect presence of leached protein-A which is co-eluted along with a huge excess of mAb and is therefore likely to be present as complex with one or several mAb molecules. To measure the exact concentration of protein-A, such complexes need to be first dissociated by boiling in the presence of SDS [28] or by employing microwave [29]. Leached protein-A and its fragments could be removed using anion exchange chromatography [27, 24 30], cation exchange chromatography [10, 30], and hydroxyapatite chromatography [18]. Some researchers have attempted to combine mAb aggregate and protein-A removal in a single step. Zhou et al. [31] showed that cation exchange chromatography using hybrid gradients of pH and conductivity resulted in removal of mAb aggregates and leached protein-A while Kelley et al. [30] used upgraded anion exchange chromatography for achieving the same objective. However, there is a limited repertoire of polishing techniques which simultaneously remove both mAb aggregates and protein-A and therefore new and efficient polishing techniques would be highly desirable. Sun et al. [32] while investigating the mechanism of IgG binding on hydrophobic surfaces, found that protein-A on its own did not interact with a hydrophobic membrane except at very high salt concentrations, i.e. protein-A was quite hydrophilic when compared with IgG. Interestingly though, IgG – protein-A complexes were found to be significantly more hydrophobic than free IgG. Any protein-A leached out from affinity media and co-eluted with mAb would, as reported by earlier workers [27, 28], be present complexed with mAb. As observed by Sun et al. [32], such complexes are likely to be more hydrophobic than the mAb. Based on this, we hypothesized that leached protein-A could be removed from mAb using hydrophobic interaction chromatography. Also, by proper optimization, removal of both mAb aggregates and protein-A could potentially be combined into a single step. 25 Figure 1 Schematic diagram for monoclonal antibody polishing based on hydrophobic interaction membrane chromatography This paper discusses a HIMC based polishing method for simultaneous removal of aggregates and leached protein-A from mAb. The working hypothesis for this method is shown in Figure 1. The feed solution for this polishing step is mAb eluted from a proteinA column using acidic buffer. In addition to mAb, the feed would therefore also be expected to contain some aggregates (represented by the dimer in the figure) and leached protein-A (which would form complex with mAb as indicated in the figure). While protein-A can bind up to five IgG molecules [33] and the figure shows just two of the possible complexed forms for the sake of simplicity. In step 1, salt was added to the feed solution to obtain conditions suitable for binding impurities on a hydrophobic membrane stack. In step 2, the mAb being less hydrophobic than the impurities was obtained in the 26 flow through while the aggregates and mAb – protein-A complexes remained bound to the membrane stack. In step 3, the membrane-bound impurities were eluted by lowering salt concentration and the membrane stack was thus regenerated. This polishing method was systematically optimized and the results obtained are discussed. 2.3 Materials and Methods 2.3.1 Materials Hydrophilized polyvinylidene fluoride (PVDF) membrane discs with 0.22 μm nominal pore diameter (catalogue no. GVWP 14250) were purchased from Millipore, Billerica, MA, USA. The average membrane thickness was 125 μm. Humanized mAb (hIgG1-CD4, batch no.10) and “dimers” (mixture of hIgG1-CD4 monomer and dimer, batch no. 6) were kindly donated by the Therapeutic Antibody Center, University of Oxford, UK. Pure hIgG1-CD4 stock solution had a total protein concentration of 4.76 mg/mL. The “dimers” stock solution had a total protein concentration of 1.29 mg/mL and contained slightly less than 50% monomer, 50% dimer and trace amounts of higher aggregates. Sodium phosphate monobasic (catalogue no. S0751), sodium phosphate dibasic (catalogue no. S0876), ammonium sulphate (catalogue no. A4418), and recombinant protein A from Staphylococcus aureus expressed in Escherichia coli (catalogue no. P7837) were purchased from Sigma-Aldrich, St. Louis, MO, USA. All buffers and test solutions were prepared using ultrapure water (18.2 MΩ-cm) water obtained from a 27 DiamondTM NANOpure water purification unit (Barnstead International, Dubuque, IA, USA). The buffers were filtered through a 0.45 μm microfiltration membrane for clarification and degassing. Protein solutions were clarified by centrifugation at 10000 revolutions per minute for 10 minutes. 2.3.2 Methods The HIMC experiments were carried using a stack of six hydrophilized PVDF membranes (18 mm effective diameter) housed in a module which was designed and fabricated in-house [34]. The module was integrated with an AKTA Prime liquid chromatography system (GE Healthcare Bioscience, Uppsala, Sweden). UV absorbance at 280nm, pH, conductivity and pressure data was collected during the chromatography experiments and logged into a computer. The feed solutions used in the HIMC experiments were adjusted to the appropriate ammonium sulphate concentration to facilitate binding. The membrane bound material was eluted with ammonium sulphate free buffer using appropriate gradients. Samples collected during these experiments were analyzed by size exclusion chromatography (SEC) and sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) [35]. SEC analysis was carried out with an Ultrahydrogel SEC column (catalogue number WAT011530, 7.8mm i.d., 30cm length, Waters, Milford, Massachusetts, USA) using an HPLC system (Prepstar 218, Varian Canada, Mississauga, Ontario). In these SEC experiments, sodium chloride solution in water (0.25 M) was used as mobile phase at a flow rate 0.3 mL/min. A 100 μL loop was used for sample injection. Reducing SDS28 PAGE (10% gel) was run using a Hoefer MiniVE vertical electrophoresis unit (GE healthcare Bio-Sciences, Montreal, Quebec, Canada). Prior to loading on the gel, samples were boiled in SDS and dithiothreitol (DTT) containing buffer to obtain individual protein subunits and to break the disulfide bonds. The electrophoresis experiments were run with normal polarity at 120 V (~35 mA current). Protein bands on the gel were visualized by staining with Coomassie blue dye. 2.4 Results and Discussion Figure 2 Effect of salt concentration on the binding of hIgG1-CD4 on hydrophilized PVDF membrane (eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + different concentrations of ammonium sulfate (1) 1.2 M, (2) 1.3 M, (3) 1.4 M and, (4) 1.5 M; hIgG-CD 4 concentration: 0.1 mg/mL, injection volume: 500 μL, flow rate: 2 mL/min) 29 Figure 2 summarizes the effect of ammonium sulphate concentration on the binding of pure mAb on a stack of six hydrophilized PVDF membrane discs. In the four experiments represented in the figure, the concentration of ammonium sulphate in the binding buffer was varied between 1.2 -1.5 M. Previous work based on the same membrane and mAb [7] had shown that the presence of 1.5 M ammonium sulphate concentration in the binding buffer resulted in strong binding of both mAb and its dimer. Therefore 1.5 M ammonium sulphate was set as the maximum salt concentration (chromatogram 1) and the salt concentration in the binding buffer was reduced in steps of 0.1 M (chromatograms 2, 3, and 4 respectively). In all four cases, elution from the membrane stack was effected by using negative salt gradient going from 0 to 100% eluting buffer, applied over 30 mL of buffer flow. As expected, in the presence of 1.5 M salt, the entire amount of mAb injected bound to the membrane stack (as indicated by the absence of a flow through peaks in chromatogram 1). The membrane-bound mAb could easily be recovered in the form of a sharp peak (51.2 mL retention volume) by switching over to the eluting buffer. However, no mAb binding took place on the membrane stack when the ammonium sulphate concentration in the binding buffer was lowered (i.e. to 1.4, 1.3 and 1.2 M, corresponding to chromatograms 2, 3 and 4 respectively) as evident from the presence of flow through peaks in these chromatograms. The shape of the flow through peak changed with change in salt concentration: being quite broad at 1.4 M, slightly sharper at 1.3 M, and very sharp at 1.2 M. This indicates that at 1.4 M salt concentration, the mAb interacted to a certain extent with the membrane stack but this was not strong enough for binding. The interaction decreased a bit at 1.3 M salt concentration and decreased even 30 further at 1.2 M salt concentration. Such change in the level of interaction is also indicated by the difference in retention volumes of the flow through peaks, these being 13.1, 6.7 and 1.5 mL respectively at 1.4, 1.3 and 1.2 M salt concentrations. Figure 3 Effect of salt concentration on the binding of hIgG1-CD4 “dimers” on hydrophilized PVDF membrane (eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + different concentrations of ammonium sulfate (1) 1.5 M, (2) 1.4 M, (3) 1.3 M and, (4) 1.2 M; hIgG1-CD 4 “dimers” concentration: 0.1 mg/mL, injection volume: 500 μL, flow rate: 2 mL/min) Figure 3 shows the effect of ammonium sulphate concentration in the binding buffer on chromatograms obtained with the “dimers”, i.e. the mAb monomer-aggregate mixture. At 1.5 M ammonium sulphate concentration, almost all components present in the injected sample bound to the membrane stack as indicated by the tiny flow through peak (see chromatogram 1). When elution was carried out using a 30 mL linear gradient form 0 to 31 100% eluting buffer, the mAb monomer and dimer were observed as sharp peaks corresponding to 50.5 and 54.1 mL retention volumes respectively. The small peaks observed after the dimer peak correspond to higher aggregates which were known to be present in small amounts in the “dimers” sample. When the binding buffer contained 1.4 M ammonium sulphate, the mAb monomer was obtained in the flow through as a very broad peak corresponding to 7.4 mL retention volume while the eluted dimer peak shifted in the forward direction to 51.4 mL (see chromatogram 2).When the salt concentration in the binding buffer was further reduced to 1.3 M, the mAb monomer was obtained as a very sharp flow through peak corresponding to a retention volume of 1.3 mL (see chromatogram 3). The reduction in the size of the dimer peak (49.7 mL retention volume) clearly indicates that some of the dimer went through the membrane stack along with the monomer without binding. This unbound dimer fraction was presumably responsible for the shoulder following the principal flow through peak. The higher aggregates were still able to bind to the membrane stack and were eluted after the dimer peak. When the ammonium sulphate concentration in the binding buffer was further reduced to 1.2 M, both mAb monomer and dimer flowed through corresponding to peaks having retention volumes of 1.3 and 3.8 mL respectively. However, the higher aggregates were still able to bind to the membrane stack and were obtained as eluted peaks (47 – 53 mL retention volume). The separation strategy for mAb polishing is to bind the mAb dimer and higher aggregates on the membrane while obtaining the mAb monomer in the flow through. The above results suggest that in order to achieve this, the ammonium sulphate concentration in the binding buffer would have to be between 1.3 and 1.4 M. The main criteria for mAb 32 polishing step is to maximize the removal of aggregates while maintaining high monomer recovery. The data shown in Figures 2 and 3 seem to suggest that the recovery of mAb would not be significantly affected by salt concentration in the above range. However, the binding capacity of aggregates could be anticipated to increase with salt concentration. Based on these considerations, 1.37 M ammonium sulphate concentration was chosen for the binding buffer for mAb polishing. Figure 4 Effect of salt concentration on the binding of protein-A on hydrophilized PVDF membrane (eluting buffer: 20 mM sodium phosphate, pH 7; binding buffer: eluting buffer + different concentrations of ammonium sulfate (1) 1.5 M, (2) 3 M; protein-A concentration: 0.1 mg/mL; injection volume: 500 μL, flow rate: 2 mL/min) Figure 4 compares the binding of protein-A on hydrophilized PVDF membrane at two different ammonium sulphate concentrations in the binding buffer, i.e. 1.5 and 3 M. At the lower salt concentration (chromatogram 1) there was no protein-A binding as evident 33 from the absence of an eluted peak. However, at 3 M salt concentration, some protein-A binding took place, the bound material being eluted as two peaks (retention volumes of 61.2 and 63.8 mL respectively). The presence of the two eluted peaks presumably indicates that some aggregation of protein-A took place at 3 M ammonium sulphate concentration. These results are consistent with those reported by Sun et al. [32], i.e. protein-A is quite hydrophilic and does not bind on a hydrophobic surface except at very high salt concentration. Table 1 Different combinations of protein-A and mAb used in HIMC experiments represented in Figure 5. mAb mass (mg) Protein-A mass (mg) Protein-A: mAb Protein-A (mass ratio) concentration (mole fraction) Chromatogram 1 0.035 0.002 0.057 0.16 Chromatogram 2 0.035 0.0015 0.043 0.125 Chromatogram 3 0.035 0.001 0.029 0.086 Chromatogram 4 0.035 0.0005 0.014 0.045 34 Figure 5 Effect of protein A amount on HIMC chromatograms obtained using mAb – protein-A mixtures (membrane: hydrophilized PVDF; eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + 1.37 M ammonium sulfate; chromatogram (1): 0.002 mg protein A + 0.035 mg mAb; chromatogram (2): 0.0015 mg protein A + 0.035 mg mAb; chromatogram (3): 0.001 mg protein A + 0.035 mg mAb; chromatogram (4): 0.0005 mg protein A + 0.035 mg mAb; injection volume: 500 μL, flow rate: 2 mL/min)* Sun et al. [32] reported that when protein-A was present in a solution along with excess rabbit IgG, it formed complexes which were more hydrophobic than the free IgG. Whether mAb –protein-A complexes behaved in a similar way was examined by carrying out HIMC experiments with different combinations of mAb and protein-A in the feed solution. The ammonium sulphate concentration in the binding buffer used in these experiments was 1.37 M. Figure 5 shows the chromatograms obtained with the different mAb – protein-A mixtures. In these experiments the amount of mAb in the sample was kept fixed while the amount of protein-A was varied. Table 1 shows the mAb and 35 protein-A contents, the mass ratio and the protein-A concentration (expressed as mole fraction) corresponding to the mixtures used to obtain chromatograms 1-4 shown in Figure 5. As the amount of protein-A in the sample was increased, the area of the flow through peak decreased and there was a corresponding increase in the amount of material eluted from the membrane stack. The reason for this is that increasing the protein-A concentration increased the amount of hydrophobic complexes in the feed solution and consequently more material bound to the membrane. These results clearly demonstrate that mAb – protein-A complexes are more hydrophobic than either mAb or protein-A, this being consistent with results obtained with protein-A and rabbit IgG mixtures [32]. The above results also show that HIMC using hydrophilized PVDF membrane stack could potentially be used to remove protein-A from mAb samples. The presence of multiple eluted peaks suggests that different types of complexes were formed. In theory one molecule of protein-A can bind up to five molecules of IgG [33]. 36 Figure 6 Polishing of mAb by hydrophobic interaction membrane chromatography (membrane: hydrophilized PVDF; eluting buffer: 20 mM sodium phosphate, pH 7.0; binding buffer: eluting buffer + 1.37 M ammonium sulfate; feed: 0.04 mg mAb monomer + 0.005 mg “dimers” + 0.0025 mg protein-A; injection volume: 500 μL, flow rate: 2 mL/min) The feasibility of polishing mAb using hydrophilized PVDF membrane was examined using simulated feed consisting of an mAb solution prepared in 1.37 M ammonium sulphate spiked with appropriate amounts of “dimers” and protein-A. The conductivity of the feed solution was 156 mS/cm and the membrane stack consisted of six discs. The injected feed sample (500 μL) which contained 0.04 mg of mAb monomer, 0.005 mg “dimers”, and 0.0025 mg of protein-A represents the worst case scenario for mAb eluted from a protein-A column. Figure 6 shows the chromatogram obtained in the above separation experiment. A 30 mL linear gradient was employed to elute membrane-bound material. The flow through peak (3.1 mL retention volume) consisted of free mAb while 37 the eluted peaks (50 – 55 mL retention volume) consisted of aggregates and mAb – protein-A complexes. Sample corresponding to the two peaks were collected and analysed by SEC and SDS-PAGE. Figure 7 Size exclusion chromatography of samples obtained from the HIMC based mAb polishing experiment (1: pure mAb monomer, 2: “dimers”, 3: simulated feed, 4: flow through, 5: eluate; column: Ultrahydrogel SEC column, mobile phase: 0.25 M sodium chloride solution in water, flow rate: 0.3 mL/min) Figure 7 shows the SEC chromatograms obtained with the mAb monomer, “dimers”, simulated feed, flow through, and eluate fractions collected during the mAb polishing experiment. The mAb monomer standard showed one peak at 28 min retention time while the “dimers” showed two peaks, the first corresponding to the mAb dimer (26 min retention time) and the second corresponding to the mAb monomer. The simulated feed solution showed two major peaks, one corresponding to the monomer, and the other 38 having a retention time of 24 min and corresponding to a much bigger molecule (approximately 450 kDa) than the dimer (approximately 300 kDa). Quite clearly, the presence of protein-A in the simulated feed resulted in the formation of large complexes containing more than one mAb molecules. While protein-A has five mAb binding sites [33], findings reported in earlier research paper seem to suggest that this theoretical maximum is rarely reached. The static binding capacity of IgG subclass 1 on protein-A has been found to be in the 3 [35] to 3.3 [36] range. The flow through obtained in the polishing experiment contained the mAb monomer only as evident from the single peak of 28 min retention time. The eluate gave two peaks, the first corresponding to the mAb – protein-A complexes and the second corresponding to the mAb monomer. The above results clearly show that while the polished mAb sample was pure, the mAb monomer could not be totally recovered in the flow through. Based on material balance, the mAb recovery was determined to be slightly above 80%. 39 Figure 8 Coomassie blue stained SDS-PAGE gel (10%, reducing) obtained with samples from the HIMC based mAb polishing experiment (M: marker; lane 1: flow through, lane 2: eluate, lane 3: simulated feed; lane 4: pure protein-A; lane 5: “dimers”; lane 6: mAb monomer) Figure 8 shows the stained SDS-PAGE gel obtained under reducing conditions with samples from the mAb polishing experiment. The molecular weight marker proteins for calibration were loaded in the “M” lane. The pure mAb monomer standard (hIgG1-CD4) was loaded in lane 6 and the “dimers” sample was loaded in lane 5. The two bands in lane 6 correspond to the heavy and light chains the mAb splits into when boiled in SDS and DTT containing buffer. The same bands were also obtained with the “dimers” sample as the aggregated forms of the mAb were first disaggregated by the action of SDS followed by their splitting into the heavy and light chains by the action of DTT. Pure protein-A was loaded in lane 4 and this gave a single band slightly higher than the 43 kDa 40 molecular marker protein. The simulated feed sample was loaded in lane 3 and this gave three bands corresponding to the heavy and light chains of mAb, and that of protein-A. The flow through sample (lane 1) gave the two signature bands of the mAb heavy and light chains while the eluate (lane 2) gave the protein-A band in addition to these. These SDS-PAGE results conclusively prove that protein-A is indeed removed by the HIMC based mAb polishing technique. The results discussed above clearly demonstrate that hydrophobic interaction membrane chromatography could be used for polishing proteinA purified monoclonal antibody. 2.5 Conclusion The experimental results discussed in this paper unambiguously demonstrate the feasibility of using hydrophobic interaction membrane chromatography for removing aggregates and leached protein-A from monoclonal antibody. The aggregates were removed by selective adsorption on the membrane as these were more hydrophobic than the monomeric form of the monoclonal antibody while protein-A was removed based on the fact that it formed relatively hydrophobic complexes with the monoclonal antibody. Under optimized solution conditions, these impurities could be simultaneously removed. The monoclonal antibody was obtained in the flow through while the impurities remained bound to the membrane and could subsequently be eluted by lowering salt concentration. 41 2.6 References [1] G. Kohler, C. Milstein, Derivation of specific antibody-producing tissue culture and tumor lines by cell fusion, Eur. J. Immunol. 6 (1976) 511-519. [2] T.A. Waldmann, Monoclonal antibodies in diagnosis and therapy, Science 252 (1991) 1657-1662. [3] P.L. Ey, S.J. Prowse, C.R. Jenkin, Isolation of pure IgG1, IgG 2a, IgG 2b immunoglobulins from mouse serum using protein A sepharose, Immunochemistry 15 (1978) 429. [4] R.L. Fahrner, D.H. Whitney, M. Vanderlaan, G.S. Blank, Performance comparison of protein A affinity-chromatography sorbents for purifying recombinant monoclonal antibodies, Biotechnol. Appl. Biochem. 30 (1999) 121128. [5] S. Hober, K. Nord, M. Linhult, Protein A chromatography for antibody purification, J. Chromatogr., B 848 (2007) 40-47. [6] J. Phillips, A. Drumm, P. Harrison, P. Bird, K. Bhamra, E. Berrie, G. Hale, Manufacture and quality control of CAMPATH-1 antibodies for clinical trials, Cytotherapy 3 (2001) 233-242. [7] L. Wang, J. Hale, R. Ghosh, Non-size-based membrane chromatographic separation and analysis of monoclonal antibody aggregates, Anal. Chem. 78 (2006) 6863-6867. [8] T. Arakawa, J.S. Philo, K. Tsumoto, R. Yumioka, D. Ejima, Elution of antibodies from a protein-A column by aqueous arginine solutions, Protein Expres Purif. 36 (2004) 244-248. 42 [9] E.M. Cromwell, E. Hilario, F. Jacobson, Protein aggregation and bioprocessing, AAPS J 8 (2006) 66. [10] J.N. Carter-Franklin, C. Victa, P. McDonald, R. Fahrner, Fragment of protein A eluted during protein A affinity chromatography, J. Chromatogr., A 1163 (2007) 105-111. [11] G.J. Ventura, A.U. Buzdar, S. Kau, B. Lichtiger, G.N. Hortobagyl, Clinical trial of plasma perfusion over immobilized Staphylococcal protein A in metastatic breast cancer, Cancer Treat. Rep. 71 (1987) 411-413. [12] K.L. Carson KL, Flexibility- the guiding principle for antibody manufacturing, Nat. Biotechnol. 23 (2005) 1054-1058. [13] S. Ghose, M. Jin, J. Liu, J. Hickey, Integrated polishing steps for monoclonal antibody purification. In: Gottschalk U, editor. Process scale purification of antibodies. New Jersey: John Wiley & Sons, Inc. New Jersey, 2009, P 145-164. [14] A. Litzen, J.K. Walter, H. Krischollek, K.G. Wahlund, Separation and quantitation of monoclonal antibody aggregates by asymmetrical flow field-flow fractionation and comparison to gel permeation chromatography, Anal. Biochem. 212 (1993) 469–480. [15] A.M. Azevedo, P.A.J. Rosa, I.F. Ferreira, M.R. Aires-Barros, Integrated process for the purification of antibodies combining aqueous two-phase extraction, hydrophobic interaction chromatography and size-exclusion chromatography, J. Chromatogr., A 1213(2008) 154-161. [16] J.P. Gabrielson, M.L. Brader, A.H. Pekar, K.B. Mathis, G. Winter, J.F. Carpenter, T.W. Randolph, Quantitation of aggregate levels in a recombinant humanized monoclonal antibody formulation by size-exclusion chromatography, 43 asymmetrical flow field flow fractionation, and sedimentation velocity, J. Pharm. Sci. 96 (2007) 268-279. [17] J. Pollastrini, Y. Jiang, Separation and characterization of protein aggregates and particles by field flow fractionation, Curr. Pharm. Biotechnol. 10 (2009) 382-90. [18] P. Gagnon, P. Ng, J. Zhen, C. Aberin, J. He, H. Mekosh, L. Cummings, S. Zaidi, R. Richieri, A ceramic hydroxyapatite-based purification platform: simultaneous removal of leached protein A, aggregates, DNA, and Endotoxins from MAbs, BioProcess Int. 4 (2006) 50-60. [19] P. Gagnon, Improved antibody aggregate removal by hydroxyapatite chromatography in the presence of polyethylene glycol, J. Immunol. Methods 336 (2008) 222-228. [20] P. McDonald, C. Victa, J.N. Carter-Franklin, R. Fahrner, Selective antibody precipitation using polyelectrolytes: A novel approach to the purification of monoclonal antibodies, Biotechnol. Bioeng. 102 (2008) 1141-1151. [21] L. Wang, R. Ghosh, Fractionation of monoclonal antibody aggregates using membrane chromatography, J. Membr. Sci. 318 (2008) 311-316. [22] Y. Lu, B. Williamson, R. Gillespie, Recent advancement in application of hydrophobic interaction chromatography for antibody aggregate removal in industrial purification process, Curr. Pharm. Biotechnol. 10 (2009) 427–433. [23] I. Kostareva, F. Hung, C. Campbell, Purification of antibody heteropolymers using hydrophobic interaction chromatography, J. Chromatogr., A 1177(2008) 254-264. [24] J. Chen, J. Tetrault, A. Ley, Comparison of standard and new generation hydrophobic interaction chromatography resins in the monoclonal antibody purification process. J. Chromatogr., A 1177 (2008) 272-281. 44 [25] M. Kuczewski, N. Fraud, R. Faber, G. Zarbis-Papastoitis, Development of a polishing step using a hydrophobic interaction membrane adsorber with a PER.C6-Drived recombinant antibody, Biotechnol. Bioeng. 105 (2010) 296-305. [26] J. Chen, J. Tetrault, Y. Zhang, A. Wasserman, G. Conley, M. DiLeo, E. Haimes, A.E. Nixon, A. Ley, The distinctive separation attributes of mixed-mode resins and their application in monoclonal antibody downstream purification process, J. Chromatogr., A 1217 (2010) 216-224. [27] J.W. Bloom, M.F. Wong, G. Mitra, Detection and reduction of protein A contamination in immobilized protein A purification monoclonal antibody preparations, J. Immunol. Methods 117 (1989) 83-89. [28] F. Steindl, C. Armbruster, R. Hahn, C. Armbruster, H.W.D. Katinger, A simple method to quantify staphylococcal protein A in the presence of human or animal IgG in various samples, J. Immunol. Methods 235 (2000) 61-69. [29] J. Zhu-Shimoni, F. Gunawan, A. Thomas, M. Vanderlaan, J. Stults, Trace level analysis of leached protein A in bioprocess samples without interference from the large excess of rhMAb IgG, J. Immunol. Methods 341 (2009) 59-67. [30] B.D. Kelley, S.A. Tobler, P. Brown, J.L. Coffman, R. Godavarti, T. Iskra, M. Switzer, S. Vunnum, Weak partitioning chromatography for anion exchange purification of monoclonal antibodies, Biotechnol. Bioeng. 101 (2008) 553-566. [31] J.X. Zhou, S. Dermawan, F. Solamo, G. Flynn, R. Stenson, T. Tressel, S. Guhan, pH-conductivity hybrid gradient cation-exchange chromatography for processscale monoclonal antibody purification, J. Chromatogr., A 1175 (2007) 69-80. [32] X. Sun, D. Yu, R. Ghosh, Study of hydrophobic interaction based binding of immunoglobulin G on synthetic membranes, J. Membr. Sci. 344 (2009)165-171. 45 [33] H. Hjelm, J. Sjodahl, J. Sjoquist, Immunologically active and structurally similar fragments of protein-A from Staphylococcus aureus, Eur. J. Biochem. 57 (1975) 395–403. [34] R. Ghosh, T. Wong, Effect of module design on the efficiency of membrane chromatographic separation processes, J. Membr. Sci. 281 (2006) 532 – 540. [35] S. Ghose, B. Hubbard, S.M. Carmer, Binding capacity differences for antibodies and Fc-Fusion proteins on protein A chromatographic materials. Biotechnol. Bioeng. 96 (2007) 768-779. [35] U.K. Laemmli, Cleavage of structural proteins during the assembly of the head of bacteriophage T4, Nature 227 (1970) 680–685. [36] A. Jungbauer, R. Hahn, Engineering Protein A affinity chromatography, Curr. Opin. Drug Discovery Dev. 7 (2004) 248. 46 Chapter 3 High-resolution hydrophobic interaction chromatographic separation of multi-protein mixture using paper-based composite membranes 2 2 This chapter is an edited version of the manuscript written by Seung Mi Yoo, Ryan Sungho Park, and Raja Ghosh. Ryan Sungho Park was a project student who conducted membrane fabrication and membrane characterization with different salts. Figure 3 and 4 was carried by Ryan Sungho Park. All his work was conducted under Seung Mi co-supervision. 47 3.1 Abstract Biopharmaceutical industry can benefit from usage of numerous protein separation methods. In this study, paper based poly (N-vinylcaprolactam) (PVCL) hydrogel composite membrane was developed for multi-protein separation purposes. These membranes showed environment responsiveness that bind and elute proteins depending on their hydrophobicity with variations in buffer salt concentration. Human immunoglobulin G (hIgG), human serum albumin (HSA) and insulin were adopted to demonstrate the membrane performance. The mixture of these three proteins was introduced to HIC butyl column and the composite membrane for comparison. The result indicated superior resolution via usage of the composite membrane over the HIC butyl column. Optimized operations of gradient and stepwise change of eluting buffer concentration demonstrated successful protein separation. HPLC-SEC and SDS-PAGE were implemented to analyze the fractionates. The flexibility of designing and constructing the composite membrane is able to allow the biopharmaceutical industry to utilize this method for multiple purposes such as drug delivery vehicle and polishing. 48 3.2 Introduction Protein separation in the biopharmaceutical industry is dominated by resin-based column chromatography based on the principles of hydrophobic interaction [1], ion exchange [2], affinity [3, 4], and size exclusion [5]. Because of its extensive applications in downstream processes, resin-based column chromatography has been developed with improved diversity in support matrix, ligand chemistry, and narrow pore distribution. Various techniques have been investigated for complex protein separations. Although many efforts have been made to improve the chromatographic resolution of protein separation, resin-based chromatography has a number of limitations. In this method, the separation of components primarily relies on pore diffusion, which requires sufficient interaction time between the mobile and stationary phases. The long process time, consequently, requires extra buffer consumption and creates high process costs. Moreover, the drop in pressure that occurs across the column poses a challenging issue because silica and soft hydrogels are easily deformed under the operating pressure. Membrane adsorbents have been utilized as selective barriers according to their sizes to remove products and process-related impurities in the biochemical engineering industry for the last 60 years [6, 7]. Advances in membrane science have improved the usability of adsorbents. They serve as surfaces and pores in affinity, charged, and hydrophobic/hydrophilic membranes, in addition to their traditional applications as filters and molecular weight cut-offs (MWCO) membranes. Because traditional bead-based chromatography exhibited some process limitations, membranes have been adopted as 49 alternative separation tools, later called membrane chromatography [8]. Several membranes are stacked in a module, where the membrane void space creates flow channels. Membrane chromatography has been actively investigated since the 1980s [9]. Membrane chromatography has brought several advancements in the field of biopharmaceutical engineering. The development of membrane technologies resulted in simple scaling-up procedures, lower pressure drops across the membrane cartridge, and larger loading volumes compared to column chromatography. It also resulted in great improvement in chromatographic resolution. Moreover, the convection flow-driven separation mechanism reduces processing time and consequently, buffer consumption, thereby leaving a small footprint [10]. Owing to these advantages of membrane chromatography, Sartorius commercialized regenerated cellulose membrane substrates with covalently attached phenyl ligands, called Sartobind® Phenyl, which are used for hydrophobic interaction chromatography. Pereira et al. (2010) demonstrated the purification of plasmid DNA using Sartobind® by manipulating the concentration of ammonium sulfate [11]. Methods used for the modification of membrane surfaces, such as plasma [12] and chemical treatment [13], ion beam irradiation [14, 15], physical adsorption (coating) [16– 19], and grafting [20–23] have been extensively investigated. In particular, hydrogel coating or grafting has been actively investigated for stimuli-responsive membranes that undergo phase transition when subjected to changes in temperature, pH, and salt concentration. These membranes are called “smart-membranes” because of their 50 responsive properties. PNIPAM has been actively researched for cross-linking or interpenetration on membrane substrates to make a three dimensional formation [14, 24, 25]. This hydrogel network can modify its physical and chemical properties upon encountering environmental stimuli. PVCL and PEG have also drawn interest in the field of biochemical engineering because of their superior biocompatibility and lower toxicity when compared to PNIPAM [26, 27]. PVCL hydrogel, like PNIPAM, is a thermo-responsive polymer that exhibits hydrophobic-hydrophilic phase transition above its LCST at 37 °C. It swells above its LCST and collapses below its LCST [28]. The presence of salt in the mobile phase tends to decrease the LCST of PVCL. Moreover, the degree of polymerization of Nvinylcaprolactam can also affect its LCST [29]. Under experimental operating conditions, at approximately 25 °C and high salt concentrations, PVCL exists in its collapsed state and attracts proteins for adsorption. On the other hand, under conditions of low ionic strength and temperatures above its LCST, it swells to its original state and releases the adsorbed proteins. Hydrophobic interaction chromatography involves the interaction between hydrophobic patches present on proteins and hydrophobic ligands on the stationary phase [30]. Even though it has been used for the separation of proteins for several decades, the mechanism underlying hydrophobic interaction is still controversial. The “salting-out” theory is the most widely accepted theory that explains hydrophobic interaction chromatography (HIC). According to this theory, surface residues of proteins form hydrogen bonds with 51 water molecules, resulting in the formation of a water-film around the protein, thereby preventing protein aggregation and precipitation. The ability to form hydrogen bonds varies with protein sequence and structure, as well as folding configuration. The apparent hydrophobicity of a protein depends on the nature and distribution of the amino acids on the protein surface (hydrophobic index/hydropathy). As salt is added to the aqueous solution, the protein loses the bound water molecules, which exposes it to the hydrophobic stationary phase. The initial mobile phase in HIC requires high salt concentrations to promote hydrophobic interaction between the protein and the stationary phase. Upon interaction with a salt (stimuli), a more favorable energetic state is generated, which results in a decreased Gibbs energy for the protein. Different salts increase the interaction between the protein and the hydrophobic stationary phase to different extents. The Hofmeister series ranks common ions according to their abilities to increase the strength of water tension around proteins. This chart is commonly consulted for the purification of proteins using the precipitation technique. The increment in molal surface tension by various salts is shown below in the decreasing order [31, 32]. MgCl2 > Na2SO4 > K2SO4 > (NH4)2SO4 > MgSO4 > Na2HPO4 > NaCl > LiCl > KSCN This order is only valid for water tension, and may vary for different proteins. It has been shown that anions dominantly control the hydrophobic strength of water tension. Therefore, the concentration of salt in the buffer is an important factor that requires careful consideration during a HIC experiment. 52 There have been numerous studies on the separation of protein mixtures by membrane chromatography using the principles of ion exchange chromatography (IEX) and HIC. Gerstner et al. (1992) reported that carboxymethyl (CM) and diethylaminoethyl (DEAE) membranes could be used to separate a three-component mixture using an ion-exchange moiety [33]. Castilho et al. (2002) demonstrated the purification of IgG from immobilized protein-A ligands using polymer (polyvinyl alcohol [PVA])-coated nylon membranes [34]. Yu et al. (2008) demonstrated that PEG-cellulose paper could be used to separate mAbs from a CHO cell culture broth [35]. Kostanski et al. (2008) characterized PVCL hydrogel-coated glass microfiber membranes to evaluate its environmental responsiveness under different ionic strengths. The membranes were characterized by evaluating their mass gain, infrared spectra, and media permeability. [36]. Huang et al. (2009) designed a commercial microfiltration membrane (microporous PVDF) crosslinked with a polymer (PVCL) for the separation of equine ferritin and hIgG using hydrophobic interactions [37]. Mah and Ghosh (2010) demonstrated the feasibility of using Whatman filter paper coated with PVCL polymer to make inexpensive composite membranes [17]. In a recent study, Himstedt et al. (2013) grew a PVCL polymer chain on a regenerated cellulose membrane. The grafting method that is used to layer the hydrogel on the membrane surface has a few advantages. First, the length of the polymer chains can be controlled easily. In addition, this is a well-defined polymerization method compared to random coating on the membrane substrate. The model proteins, BSA and IgG, were used to test the adsorption as well as the binding capacity of the membrane [37]. 53 In this study, we coated PVCL hydrogel on a Whatman filter paper membrane for use in stimulus-based hydrophobic interaction membrane chromatography [17]. Our preliminary results were published in 2010 [17]. We demonstrated that environmentally responsive PVCL composite membranes have a lower critical solution temperature in the presence of ammonium sulfate. On the basis of these results, we hypothesized that a paper-based composite membrane might be able to fractionate a multi-protein mixture by hydrophobic interaction membrane chromatography. After consulting the Hofmeister series, ammonium sulfate and sodium chloride were tested for their ability to influence membrane responsiveness. This paper discusses the fractionation of plasma proteins, HSA, hIgG, and a popular hormone, insulin, using a “custom made” PVCL composite membrane chromatography system based on hydrophobic interactions. HSA and hIgG are the two most abundant proteins in human blood plasma and occur in a ratio of 7:2. In addition, insulin is an important molecule in the pharmaceutical industry. Furthermore, the differences in their hydrophobicities are very subtle [38]. To demonstrate the large capacity of the composite membranes, bioseparation was tested for a large amount of protein. These membranes allowed high flow rates while maintaining relatively low pressures. Moreover, they had a high selectivity for proteins. Fractionated proteins were collected and analyzed via high performance liquid chromatography-size exclusion chromatography (HPLC-SEC) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The present 54 work aims to demonstrate the bioseparation of a multi-protein mixture and address some challenges that are encountered when using paper-based composite membranes. 3.3 Materials and Methods 3.3.1 Materials Whatman filter paper purchased (no.5, catalogue # 28462-166) from VWR and was used for support matrix on composite membrane. Monomer, N-vinylcaprolactam (catalogue #415464), and cross-linker, methylene-bisacrylamide (catalogue #146072), UV-initiator, diphenyl (2,4,6-trimethylbenzoyl)-phosphine oxide/2-hydroxy-2-methylpropiophenone, catalogue #405663) was purchased from Sigma-Aldrich, St. Louise, MO, USA. The HPLC-grad n-isopropanol (catalogue # AH323-4) was purchased from Fisher Scientific, Ottawa, ON, Canada. Human immunoglobulin G (hIgG) (≥95% purity, catalogue #14506), Insulin from bovine pancreas (catalogue number I6634), sodium phosphate (mono- and dibasic) (catalogue # S0751 and S0876), ammonium sulphate (catalogue #A4418), and sodium chloride (catalogue # S5886) were purchased from Sigma-Aldrich, St. Louise, MO, USA. Purified human serum albumin (HSA) was kindly donated by the Scottish Blood Transfusion Service, Edinburgh, UK. All buffers and protein solutions were prepared using ultrapure water (18.2 MΩ-cm) obtained from Diamond NANOpure water purification unit (Barnstead International, Dubuque, IA, USA) and were filtered through a 0.45 μm microfiltration membrane for clearing and degassing. 55 3.3.2 Separation of protein mixture with HIC butyl column The protein mixture was consisted with human immunoglobulin, serum albumin, and insulin with the mass ratio of 1.5: 2.5: 1. The protein mixture was loaded at the flow rate of 1 mL/min onto a 1 mL column of butyl HIC (GE healthcare bioscience, Uppsala, Sweden) where integrated with AKTA prime liquid chromatography system (GE healthcare bioscience, Uppsala, Sweden). This process was obtained for comparison of protein resolutions. The protein sample loading condition was equilibrated in 1.5 or 1.3 M ammonium sulphate buffer (pH 7) composed of 20 mM of sodium phosphate. The loading condition was determined by optimization of individual protein bindings prior. After protein mixture injected into AKTA prime system along with binding buffer, stabilization time has given. Once the chromatogram became the plateau, a linear gradient of decreasing ammonium sulphate concentration (1.5 or 1.3 to 1 M) was applied over 20 mL of sodium phosphate. All the flow through and eluting proteins were collected for further protein analysis. The desalting and concentrating of those fractions was conducted with Amicon ultra centrifugal (3 kDa NMWCO) filter devices (Millipore Corporation, Bedford, MA). 3.3.3 Paper based composite membrane preparation The Whatman filter paper were cut in 20 mm diameter discs and conditioned for measuring the mass of paper discs in constant temperature and humidity (23 °C and 50 % relative humidity) room. The polymer solution was prepared in 3g of n-isopropanol with different molar ratio of monomer (30%, 35% and 40%) and cross-linkers (2%, 3%, and 56 4%) and 0.2 of UV-initiator; the recipes were adopted from Mah and Ghosh 2010 [17]. The conditioned filter papers were immersed into the coating (polymer) solution for 5 min. The individual discs were sandwiched within laminating pouches and pressed with rollers to seal filter paper discs entrapped polymer solution. This process also eliminated any possible of oxygen inhibition on polymerization and well distribution of polymer solution within membrane pores. The sealed discs were placed in UV chamber with UVirradiation at 350-400 nm for 20 min (3.3 mW/cm2 intensity). After fully polymerized membrane discs were removed from laminating pouches and rinsed with DI water in agitator at 37 °C for 24 hours. The composite membrane discs were weighed after dried in paper conditioning room (23 °C and 50 % relative humidity). The mass gain and thickness of composite membrane were measured with 20 discs. 3.3.4 Membrane chromatography set-up Five same type composite membranes (20-mm diameter) were stacked within a customdesigned membrane module [17] which was then integrated with an AKTA prime liquid chromatography system (GE Healthcare Biosciences, BAIE D'urfe, QC, Canada). The UV absorbance (at 280 nm), conductivity and pH of the effluent stream from the module as well as the system pressure data were logged into a computer using Prime View. Appropriate loops were used to inject the feed samples into the membrane module. All membrane chromatography experiments were carried out at ambient temperature (24 °C). 57 3.3.5 Hydraulic testing of membranes Membrane stacks were tested for hydraulic permeability at 1 mL/min flow rate using two buffers: 20 mM sodium phosphate pH 7.0 (buffer B) and 1.1 M ammonium sulphate prepared in 20 mM sodium phosphate pH 7.0 (buffer A). The flow of buffers A and B through the membrane stack was alternated several times and the backpressure was recorded. 3.3.6 SDS-PAGE Non-reducing SDS-PAGE (10% gel) was run by Hoefer MiniVE vertical electrophoresis unit (GE healthcare Bioscience, Montreal, Quebec, Canada). Prior to loading on the gel, samples were boiled in SDS containing buffer. A constant current 25mA/gel for 1.5 h in separating gel was applied [39]. Protein bands on the gel were stained with 0.1% Coomassie Brilliant Blue R-250 for 30 min at room temperature, followed by destaining in 10 % (v/v) acetic acid and 30 % (v/v) methanol in water. The gel picture was obtained using Bio-Imaging MiniBis Pto system (24-25-PR) purchased from DNR-Imaging System, Jerusalem, Israel. 3.3.7 SEC-HPLC SEC analysis was carried out with a TSKgel G4000 SWXL column (catalogue number 08542, 7.8 mm i.d., 30 cm length, TOSOH Bioscience LLC, King of Prussia, PA, USA) using HPLC system (Prepstar 218, Varian Canada, Missussuage, Ontario). In these SEC experiments, mobile phase; 0.25M of sodium chloride in 20 mM of sodium phosphate solution at pH 7, was carried out at a flow rate of 0.5mL/min. Each analysis was injected 58 with 50 μL volume. The mobile phase was filtered through a 0.1 μm microfiltration membrane for clearing and degassing. 3.4 Results and Discussions 3.4.1 Optimization of protein separation with resin based hydrophobic interaction chromatography Figure 1 Effect of salt concentration on the binding of three proteins (hIgG, HSA, and Insulin) on butyl Sepharose TM HIC (eluting buffer: 20mM sodium phosphate, pH 7; binding buffer: eluting buffer+ different concentrations of ammonium sulphate – (1) 1.3 M, (2) 1.5 M; model protein concentration: hIgG 0.21 mg+ HSA 0.35 mg+ Insulin 0.14 mg in 500μL injection volume) 59 The suitability of the membranes as chromatographic support had to be confirmed by assessing their performance by challenging with the same proteins binding and eluting condition on both bead-based HIC butyl column and membranes. The first chromatographic runs were done by using pH 7.0, 1.3 M ammonium sulphate in 20 mM sodium phosphate as the binding buffer and pH 7.0, 20 mM sodium phosphate as the elution buffer. This is illustrated in Figure 1. The elution was done by negative gradient over 20mL of buffer flow on HIC butyl bead-based column. The injected protein solution was previously conditioned by adding ammonium sulphate salt up to 1.3M. The second chromatographic runs were done by identical chromatographic conditions except the binding buffer was 1.5 M ammonium sulphate. The maximum binding condition was limited by insulin aggregation; at ammonium sulphate concentration of 1.5 M, the insulin protein solution began to become cloudy which can induce negative effects on the hydrophobicity of the interaction between the protein and butyl column. Higher concentration of lyotropic salt takes up water molecules and consequently insulin protein molecules form dimer to hexamers (reversible aggregates)[35]. The injection protein sample (500 μL) containing 0.21 mg of hIgG, 0.35 mg of HSA, and 0.14 mg of insulin; accounting to a total protein amount of 0.7 mg per 1 mL of HIC butyl column bed volume. Both of chromatogram 1 and 2 had flow through peaks (1-2 mL retention volume) and elution peaks (11-25 mL retention volume) obtained by negative salt concentration. As expected, chromatogram 2 has smaller flow through protein peak while more protein was eluted at 11-25 mL effluent volume since higher salt concentration induced more hydrophobic interaction between proteins and stationary phase (butyl 60 column). The flow through peak (peak a) and elute peaks (peak b and c) samples in the case of 1.3 M salt concentration (chromatogram 1) were collected for SDS-PAGE analysis. Figure 2 Coomassie blue stained SDS-PAGE gel (10%, non-reducing) obtained with samples from the butyl Sepharose TM HIC. Multi-protein mixture (M: marker; lane 1: hIgG, lane 2: HSA, lane 3: insulin, lane 4: feed –three protein mixture, lane 5: flow through-peak a, lane 6: first elute-peak b, lane 7: second elute-peak c) The feed, flow through and eluted protein samples were analyzed by 10 % non-reducing SDS-PAGE, the results were shown in Figure 2. The molecular marker loaded “M” lane for reference and three standard samples were loaded in lane 1 (hIgG), lane 2 (HSA), and lane 3 (insulin). The hIgG (150 kDa) had a band on the top of gel while HSA has two bands at 150 kDa and 67 kDa due to the traceable amount of hIgG present in plasma stock solution. The molecular weight of insulin is 5.8 kDa which appeared below 10 kDa 61 protein marker. In the figure, the color of the insulin band was not clearly shown; it had silver color while other proteins were stained by Coomassie blue. Because insulin naturally contained metal (Zn), its band on the gel is not blue. The feed sample (mixture of three proteins), the collected samples (peak a, b, and c) were loaded in lane 4, 5, 6 and 7 respectively. For the feed sample (lane 4), three bands were observed corresponding to hIgG, HSA, and insulin. The gel picture for the flow through (lane 5) fraction of the butyl HIC experiment showed two faint bands which corresponded to the hIgG and HSA respectively. The first eluate (peak b) showed a similar band as the flow through fraction with a darker band while second eluate (peak c) contained all three proteins (hIgG, HSA, and insulin). It clearly showed separation of these three proteins with HIC butyl column is not the best choice. 62 3.4.2 Composite paper based membrane characterizations (salt responsive and protein binding/elution test) Figure 3 Pressure profile in buffer change experiment carried out using filter paper based composite membranes (flow rate: 1.0 mL/min; 1.5 M ammonium sulphate in 20 mM sodium phosphate buffer, 3M of sodium chloride in 20 mM sodium phosphate buffer, pH 7.0) Based on previous work [17], among the 5 recipes of composite membranes, a composition of 40 mass % monomer and 2 mole % cross-linker was selected for protein separation experiment due to the reasonable hydraulic permeability (2.55 X 10-6 m Pa-1 s1 per m of bed height) and the saturation hIgG binding capacity (12 mg/mL bed volume) at 1.1M ammonium sulphate. The five-membrane stack was placed in customized membrane module and its transmembrane pressure (ΔP) was tested under two different salts as shown in Figure 3. One salt is ammonium sulphate and the other is sodium chloride which was intended for the potential usage on biological system application. 63 Since the PVCL hydrogel coating on filter paper has lower critical solution temperature (LCST) of 32 °C, the polymer layer collapses in a high ionic strength solution when operated at room temperature [41, 42]. Salt induces the hydrogel to collapse from swollen salts as it explains in introduction section. It also causes the increase in the void space within membrane pores and shortens tortuosity. The transmembrane pressure is a good indication of the hydrogel state as well as hydrophobicity of membrane surface. As expected membrane back pressure was as high as 0.046 MPa (ammonium sulphate) and 0.038 MPa with 2.5 X 10-3 (Ns/m2) (sodium chloride) shown in Figure 3. As more salt was introduced to the system as conductivity indicated, the membrane back pressure dramatically decreased to approximately 0.035 MPa (in both salt cases) since the hydrogel was collapsed. Under salt free environment, PVCL polymers are swollen to induced protein desorption mode. On the other hand, when the salt is present, PVCL polymer can collapse to the cellulose filter papers; in the collapsed condition more favorable for protein adsorption. Regardless of the responsive properties of PVCL, proteins are able to pass through the membrane because the pore size is much bigger than the protein hydrodynamic radii. In contrast, non-coated membrane (Whatman filter paper itself) has no environment response by salt additives. As the salt concentration increases, the viscosity of mobile phase also increases (higher salt buffer cause higher transmembrane pressure due to the salt liquid viscosity). For example, sodium chloride solution has dynamic viscosity difference between 1 to 2.5 X10-3 N s/m2 at 0-3 M concentration. The environmental responsiveness was manipulated for multi-component 64 separation with membrane chromatography [17,19,43]. Most previous works were done only with ammonium sulphate solution. However, in addition to ammonium sulphate, sodium chloride solution was also used in this study. The membranes are responsive to sodium chloride as well although the pressure difference is not as high as ammonium sulphate solution. This requires further investigation of appropriate model protein with strong hydrophobicity also not for separation applications (not shown in this dissertation). Figure 4 Protein binding effect test using composite membrane, hIgG 0.5mg, and insulin 0.3 mg in 500 μL injection loop(flow rate: 1.0 mL/min; 0.6M and 0.8M of ammonium sulphate in 20mM sodium phosphate buffer, pH 7.0) Individual protein (hIgG and insulin) binding and elution was studied under different ammonium sulphate concentration as shown in Figure 4. Insulin (0.3mg) was loaded at 65 0.6 M of ammonium sulphate as binding condition; however, UV absorbance indicated unbound insulin flowed through the membrane system which indicates it was not hydrophobic enough for insulin molecules. The second chromatogram indicates 0.8 M of ammonium sulphate is strong enough for insulin and membrane interactions, and after elution buffer applied to the system, insulin eluted as shown in Figure 4a. Similarly, hydrophobicity of hIgG (0.5mg) was tested at 0.6M and 0.8Mammonium sulphate binding condition. The protein hydropathy index is commonly used as a reference for estimation of proteins and their stationary interactions. Each amino acid has hydrophobic scale and summation of each numbers indicates hydrophobicity/hydrophilicity of proteins. According to the hydropathy index, all three proteins have values below zero which means they are hydrophobic. However, there are limitations for assessing degrees of hydrophobicity scale on each protein, because they have three dimensional structures in the liquid solution [44]. Based on individual protein binding experiment as shown in Figure 4, three protein mixtures were investigated on fractionations with paper based composite membrane using hydrophobic interactions. 66 3.4.3 Process optimization (elution mode) Figure 5 Effect of salt concentration on the binding of three model proteins on PVCL composite membrane with 20 mL of gradient (eluting buffer: 20 mm sodium phosphate, pH 7.0; binding suffer: eluting buffer + 1.3 M ammonium sulphate, injected proteins concentration: IgG 0.21 mg+ HSA 0.35 mg+ Insulin 0.14 mg in 500μL injection volume, total protein loading 0.7 mg/0.25 mL of bed volume, flow rate: 1.0 mL/min) Salt-responsive membrane chromatography was applied to the separation of proteins. As shown in Figure 5, the separation of a mixture of three proteins, hIgG (0.21 mg) and HSA (0.35 mg) and insulin (0.14 mg) was achieved by changing the salt concentration. The total protein mass loaded was 0.7 mg in 0.25 mL membrane stack volume. The saturation binding capacity of hIgG at 1.1 M of ammonium sulphate was 12 mg/mL [17]. The binding buffer consists of 20 mM sodium phosphate (pH 7.0) containing 1.3 M of ammonium sulphate. This solution passed through the system containing the membrane 67 stack. According to Figure 4, under 1.3 M of ammonium sulphate concentration, both hIgG and insulin are bound to the membrane stack while HSA has no interactions with the membrane. Elution was carried out by a low salt condition which was 20 mM sodium phosphate for 20 mL of linear gradient. The elution order of the three proteins reflects their hydrophobic properties; less hydrophobic proteins are eluted earlier and more hydrophobic proteins are eluted from membrane later. Similar study was done with hydrophobic interaction column chromatography (HIC) in order to verify the protein hydrophobicity by its retention time [38]. As discussed in section 3.2, the property of the surface of the stationary phase (PVCL coated membrane) was altered from hydrophobic to hydrophilic. At effluent volume of 1-8 mL, the flow through peak was observed, and the two elution peaks were observed at 18-28 mL and 28-38 mL of effluent volume. When compared to other commercialized membranes (i.e. PVDF[43]),the resolution of chromatogram had protein release for a longer period of time. The samples in accordance with the peaks (FT, E1 and E2) were collected, and analysed for their protein contents by SEC-HPLC and SDS-PAGE. 68 Figure 6 Size exclusion chromatography of samples obtained from the HIMC based multi-proteins separation a) pure standard samples 1: hIgG, 2: HSA, 3: Insulin, 4: feed – (hIgG+ HSA+ Insulin), b) fractions from figure 5, 1: flow through, 2: first eluting peak 3: second eluting peak; column: TSKgel G3000SWXL, mobile phase: 0.25 M sodium chloride solution in 20 mM of sodium phosphate, flow rate: 0.3 mL/min) Figure 6 shows the SEC chromatography obtained from standard samples and fractions from the gradient mode membrane separation. The standard sample chromatograms shows that the retention times of hIgG, HSA, and insulin were 13.5 min, 15 min, and 26 min respectively. The chromatogram for the feed solution showed three peaks, fist shoulder corresponded to hIgG, while the second peak had a retention time at 15 min which corresponded to HSA, and the broad peak at 26 min corresponded to insulin. The flow through and the two elute peaks were collected during the 20 mL gradient separation condition. The flow through peak in the separation experiment contained the HSA; this 69 was shown by SEC analysis with a single peak of 15 min retention time. The first elute peak showed one peak in SEC analysis at 13 min indicating only hIgG was present; while in the second elute fraction, two small and one large peak at 13 min, 15 min, and 26 min were observed. Therefore, by using gradient elution, HSA can be resolved in flow through while hIgG can be collected in first eluted peak. However, the second elute peak contained some residual hIgG and HSA remained with insulin. Figure 7 Coomassie blue stained SDS-PAGE gel (10%, reducing) obtained with samples from the HIMC based multi-protein mixture (M: marker; lane 1: hIgG, lane 2: HSA, lane 3: insulin, lane 4: feed –three protein mixture, lane 5: flow through, lane 6: first elute peak, lane 7: second elute peak The flow through and eluate protein fractions from the PVCL coated membrane chromatography separation were analyzed with 10 % non-reducing PAGE. Figure 7 shows the flow through and two eluates collected from 20 mL gradient elution mode. The 70 molecular marker was loaded at “M” lane for reference and three standard samples were loaded in lane 1 (hIgG), lane 2 (HSA), and lane 3 (insulin). The feed sample (mixture of three proteins) and the three fractions were loaded in lane 4, 5, 6 and 7 respectively. The PAGE gel was more sensitive than SEC since HSA in SEC data did not show hIgG peak. The multi-protein feed sample was loaded in lane 4, which had three bands on the gel corresponded to hIgG, HSA, and insulin respectively. Lane 5 contained the flow through; faint hIgG and dark broad HSA bands were observed. Lane 6 was loaded with the first eluate sample (peak E1) which mostly contained hIgG. The second eluate (lane 7) was analyzed and it seemed to have a small amount of hIgG though the primary content was insulin. Same as the SEC analysis data, with gradient mode elution, trace amount of unseparated proteins were still observed in fractions. This indicated there are still rooms for improvement in resolution of protein separation. 71 Figure 8 Effect of salt concentration on the binding of three model proteins on PVCL composite membrane with step change 0-70-100 % (eluting buffer: 20 mm sodium phosphate, pH 7.0; binding suffer: eluting buffer + 1.3 M ammonium sulphate, injected proteins concentration: IgG 0.21 mg+ HSA 0.35 mg+ Insulin 0.14 mg in 500μL injection volume, flow rate: 1.0 mL/min) The attempt to separate multi-proteins using PVCL composite membrane with gradient operation mode has been described in section 3.3.1. In the following sections, the elution was carried out by step change of buffer salt concentration for comparison. Figure 8 showed multi-protein separation using coated membranes with 1.3 M binding buffer and the elution was carried out by step change of elution buffer; 0 to 70 % and 70 to 100% of 20 mM of phosphate. The flow through (FT) peak was shown at 1-8 mL of retention volume and fist elution peak (E1) was collected at 20 to 26 mL. The second elution peak (E2) appeared as a broad shoulder peak from 32 to 38 mL. As expected, the shapes of the peaks were sharper than the ones from gradient elution experiment due to the PVCL 72 hydrogel phase transition by sudden salt removal. All three fractions were collected for further protein analysis, HPLC-SEC and SDS-PAGE. Figure 9 Size exclusion chromatography of samples obtained from the HIMC based multi-proteins separation a) pure standard samples 1: hIgG, 2: HSA, 3: Insulin, 4: feed – (hIgG+ HSA+ Insulin), b) fractions from figure 8, 1: flow through, 2: first elute peak, 3: second elute peak, column: TSKgel G3000SWXL, mobile phase: 0.25 M sodium chloride solution in 20 mM of sodium phosphate, flow rate: 0.3 mL/min) Figure 9 represents the obtained chromatograms, for the flow through, and the two elute fractions collected during the hydrophobic interaction membrane chromatography (HIMC) stepwise (0-70-100 % of eluting buffer) separation. The flow through fraction chromatogram (FT) has a single peak at 15 min which corresponded to HSA, and one peak was observed for the first eluate fractionation (E1) at 13 min which corresponded to 73 hIgG. The last chromatogram, which was for the second eluate (E2), also had only one peak at 26 min indicating only insulin was present in this fraction. This was strong evidence showing that HSA protein (among three proteins) was able to flow through the hydrogel coated membrane chromatography completely without any interaction with the membrane since there is no HSA observed in any of the eluate fraction; meanwhile, hIgG and insulin were bound to the membrane until the change in environment to lower salt concentration. The hIgG’s elution was followed by insulin in the stepwise elution. Comparing with the linear gradient mode, the stepwise elution has a better resolution as shown in SEC-HPLC analysis. Figure 10 Coomassie blue stained SDS-PAGE gel (10%, reducing) obtained with samples from Figure 8 (M: marker; lane 1: hIgG, lane 2: HSA, lane 3: insulin, lane 4: feed –three protein mixture, lane 5: flow through, lane 6: first elute peak, lane 7: second elute peak) 74 The fraction samples collected from stepwise separation experiment were analysed by SDS-PAGE, and the results are shown in Figure 10. Reference molecular ladder was loaded in lane “M” and standard protein samples were loaded in lane 1 (hIgG), lane 2 (HSA), and lane 3 (insulin). The feed (mixture of three) sample was loaded in lane 4. Lane 5 showed the flow through (FT) fraction which was collected from stepwise separation only for the HSA content. While lane 6 (E1) showed primarily hIgG which was collected from the step change of 0 – 70 % elution buffer. Finally, lane 7 (E2) indicated the primary content in the second elution peak was insulin with a trace amount of hIgG, and this was collected from the step change of 70 – 100 % elution buffer. Compared with the separation of hIgG, HSA, and insulin using gradient elution, the resolution of stepwise elution improved significantly. However, in real pharmaceutical industry, it is challenging to operate a sharp change in salt concentration; it is likely that the linear gradient condition is more suitable for industry scale operation. This research project showed the feasibility of optimized operating condition; it also showed that PVCL coated membrane was able to separate multi-component proteins. 3.5 Conclusions Hydrophobic interaction membrane chromatography has been actively developed for performance and cost reduced membrane. Here, poly (N-vinylcaprolactam) (PVCL) hydrogel was coated on filter paper to obtain a responsive membrane since PVCL hydrogel is LCST polymer. Lyotropic salts were lowering LCST at the same time being a 75 trigger for phase transition. As more ionic strength applies to membrane environment, PVCL polymer deformed and collapsed. By manipulating responsive properties, switchable membrane hydrophobicity (physical and chemical properties) successfully demonstrated for three proteins separation. HIC butyl column was tested along with the composite membrane in order to compare the resolution. The least hydrophobic protein (HSA) flowed through at 20 mM of sodium phosphate in 1.3 M of ammonium sulphate. The second most hydrophobic protein (hIgG) was eluted after 70% of stepwise elution buffer obtained to the membrane chromatography system. Lastly, the most hydrophobic protein (insulin) was eluted when 100 % of eluting buffer applied in the membrane chromatography which the responsive membrane undergoes hydrated state. Optimized elution mode which is stepwise change in elution buffer concentration, had the best resolution as visualized in HPLC-SEC and SDS-PAGE protein analysis. The composite membrane experienced some of fouling problems, and less sensitive phase transition than grafted commercialized membrane (i.e. hydrophilized PVDF) [43]. Nevertheless, the composite membrane has more freedom to tailor with degree of hydrogel crosslinking and membrane size or shape, and higher permeability good for mass process unit such as waste water treatment [45]. Product driven design is necessary depending on the applications. For example, hydrophobic protein interaction for drug delivery vehicle requires high content of hydrogel. On the other hand, hydraulic permeate flux achievement in polishing step can implement less hydrogel content membranes. This cost effective development of hydrogel based membrane chromatography could significantly benefit the downstream purification or polishing step in biopharmaceutical industry. 76 3.6 References [1] S. Hierten, Some general aspects of hydrophobic interaction chromatography, J. Chromatogr. A. 87 (1973) 325-331. [2] P.R. Levison, C. Mumford, M. Streater, A. Brandt-Nielsen, N.D. Pathirana, S.E. Badger, Performance comparison of low-pressure ion-exchange chromatography media for protein separation, J. Chromatogr. A. 760 (1997) 151–158. [3] H. Lakhiari, D. Muller, Purification of IgG and insulin on supports grafted by sialic acid developing “thiophilic-like” interactions, J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 818 (2005) 53–9. [4] R. Hahn, R. Schlegel, A. Jungbauer, Comparison of protein A affinity sorbents, 790 (2003) 35–51. [5] J. Porath, P. Flodin, Gel filtration: A mrthod for desalting and group separation, Nature. 183 (1959) 1657–1659. [6] R.W. Baker, Membrane technology. Encyclopedia of polymer science and technology, John Wiley & Sons, Inc., Membrane technology & research, Inc., Menlo park, California, 2001. [7] D.K. Roper, E.N. Lightfoot, Separation of biomolecules using adsorptive membranes, J. Chromatogr. A. 702 (1995) 3–26. [8] R. van Reis, a Zydney, Membrane separations in biotechnology., Curr. Opin. Biotechnol. 12 (2001) 208–11. [9] S.R. Eisenberg, A.J. Grodzinsky, Electrically modulated membrane permeability, J. Memb. Sci. 19 (1984) 173–194. [10] R. Ghosh, Protein separation using membrane chromatography: opportunities and challenges., J. Chromatogr. A. 952 (2002) 13–27. [11] L.R. Pereira, D.M.F. Prazeres, M. Mateus, Hydrophobic interaction membrane chromatography for plasmid DNA purification: Design and optimization., J. Sep. Sci. 33 (2010) 1175–84. [12] K. Asfardjani, Y. Secui, Y. Aurelle, N. Abidine, Effect of Plasma Treatments on Wettability of Polysulfone and Polyetherimide, 43 (1991) 271–281. 77 [13] D. Mukherjee, A. Kulkarni, W.N. Gill, Chemical treatment for improved performance of reverse osmosis membranes, 104 (1996) 239–249. [14] R. Chennamsetty, I. Escobar, X. Xu, Characterization of commercial water treatment membranes modified via ion beam irradiation, Desalination. 188 (2006) 203–212. [15] K. Good, I. Escobar, X. Xu, M. Coleman, M. Ponting, Modification of commercial water treatment membranes by ion beam irradiation, Desalination. 146 (2002) 259–264. [16] J. Lei, M. Ulbricht, Macroinitiator-mediated photoreactive coating of membrane surfaces with antifouling hydrogel layers, J. Memb. Sci. 455 (2014) 207–218. [17] K.Z. Mah, R. Ghosh, Paper-based composite lyotropic salt-responsive membranes for chromatographic separation of proteins, J. Memb. Sci. 360 (2010) 149–154. [18] R. Huang, K.Z. Mah, M. Malta, L.K. Kostanski, C.D.M. Filipe, R. Ghosh, Chromatographic separation of proteins using hydrophobic membrane shielded with an environment-responsive hydrogel, J. Memb. Sci. 345 (2009) 177–182. [19] R. Huang, L.K. Kostanski, C.D.M. Filipe, R. Ghosh, Environment-responsive hydrogel-based ultrafiltration membranes for protein bioseparation, J. Memb. Sci. 336 (2009) 42–49. [20] J.E. Kilduff, S. Mattaraj, M. Zhou, G. Belfort, Kinetics of Membrane Flux Decline: The Role of Natural Colloids and Mitigation via Membrane Surface Modification, J. Nanoparticle Res. 7 (2005) 525–544. [21] N. Hilal, V. Kochkodan, L. Al-Khatib, T. Levadna, Surface modified polymeric membranes to reduce (bio)fouling: a microbiological study using E. coli, Desalination. 167 (2004) 293–300. [22] J. Pieracci, J. V Crivello, G. Belfort, Photochemical modification of 10 kDa polyethersulfone ultrafltration membranes for reduction of biofouling, J. Memb. Sci. 156 (1999) 223–240. [23] M. Taniguchi, G. Belfort, Low protein fouling synthetic membranes by UVassisted surface grafting modification: varying monomer type, J. Memb. Sci. 231 (2004) 147–157. 78 [24] M. Rossi, D. Crespy, Temperature-responsive polymers with LCST in the physiological range and their applications in textiles TEMPERATURE IN THE PHYSIOLOGICAL, Polym. Int. 1468 (2007) 1461–1468. [25] G.V.R. Rao, M.E. Krug, S. Balamurugan, H. Xu, Q. Xu, G.P. Lo, Synthesis and Characterization of Silica - Poly ( N -isopropylacrylamide ) Hybrid Membranes : Switchable Molecular Filters, Chem. Mater. (2002) 5075–5080. [26] H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu, J. Hirvonen, Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam), Biomaterials. 26 (2005) 3055–3064. [27] J. Kopecek, Hydrogel biomaterials: a smart future?, Biomaterials. 28 (2007) 5185– 92. [28] M. Prabaharan, J.J. Grailer, D.A. Steeber, S. Gong, Stimuli-responsive chitosangraft-poly (N-vinylcaprolactam) as a promising material for controlled hydrophobic drug delivery, Macromol. Biosci., 8 (2008), 843–851 [29] N.S. Ieong, M. Hasan, D.J. Phillips, Y. Saaka, R.K. O’Reilly, M.I. Gibson, Polymers with molecular weight dependent LCSTs are essential for cooperative behaviour, Polym. Chem., 3 (2012) 794–799. [30] W.R. Melander, D. Corradini, C. Horvath, Salt-mediated retention of proteins in hydrophobic-interaction chromatography. Application of solvophobic theory, J Chromatogr, 317 (1984) 67-85. [31] Y. Zhang, P.S. Cremer, Interactions between macromolecules and ions: the Hofmeister series, Curr Opin, Chem, Biol, 10 (2006) 658-663. [32] F. Hofmeister, Zur lehre von der wirkung der salze. Naunyn-Schmiedeberg’s Archives of Pharmacology 24 (1888) 247-260. [33] J.A. Gerstner, R. Hamilton, S.M. Cramer, Membrane chromatographic protein separations systems for high-throughput, 596 (1992) 173–180. [34] L.R. Castilho, F.B. Anspach, W.-D. Deckwer, Comparison of affinity membranes for the purification of immunoglobulins, J. Memb. Sci. 207 (2002) 253–264. [35] D. Yu, X. Chen, R. Pelton, R. Ghosh, Paper-PEG-based membranes for hydrophobic interaction chromatography: purification of monoclonal antibody., Biotechnol. Bioeng. 99 (2008) 1434–42. 79 [36] L.K. Kostanski, R. Huang, R. Ghosh, C.D.M. Filipe, Biocompatible poly(Nvinyllactam)-based materials with environmentally-responsive permeability., J. Biomater. Sci. Polym. Ed. 19 (2008) 275–90. [37] H.H. Himstedt, X. Qian, J.R. Weaver, S.R. Wickramasinghe, Responsive membranes for hydrophobic interaction chromatography, J. Memb. Sci. 447 (2013) 335–344. [38] A. Ladiwala, F. Xia, Q. Luo, C.M. Breneman, S.M. Cramer, Investigation of protein retention and selectivity in HIC systems using quantitative structure retention relationship models., Biotechnol. Bioeng. 93 (2006) 836–50. [39] U.K. Laemmli, Cleavage of structural proteins during the assembly of the head of bacteriophage T4, Nature. 227 (1970) 680–685. [40] W. Wang, Protein aggregation and its inhibition in biopharmaceutics., Int. J. Pharm. 289 (2005) 1–30. [41] S. Saeki, N. Kuwahara, M. Nakata, M. Kaneko, Phase separation of poly (ethylene glycol) -water-salt systems, 18 (1977) 1027–1031. [42] S. Peng, C. Wu, Surfactant Effect on pH and Temperature Sensitivities of Poly ( N -vinylcaprolactam- co -sodium acrylate ) Microgels, (2001) 568–571. [43] S.M. Yoo, R. Ghosh, Simultaneous removal of leached protein-A and aggregates from monoclonal antibody using hydrophobic interaction membrane chromatography, J. Memb. Sci. 390-391 (2012) 263–269. [44] A. Mahn, M.E. Lienqueo, J.C. Salgado, Methods of calculating protein hydrophobicity and their application in developing correlations to predict hydrophobic interaction chromatography retention., J. Chromatogr. A. 1216 (2009) 1838–1844. [45] E.G. Mah, Fabrication of paper based thermo-responsive membranes and investigation for their use in adsorption of emerging water contaminants, McMaster dissertation, 2012. 80 Chapter 4 Coating the surface of hollow fiber membranes with a natural hydrogel for use in bioreactors3 3 This chapter is an edited version of a manuscript written by Seung Mi Yoo and Raja Ghosh. This manuscript is ready to journal submission. 81 4.1 Abstract The use of hollow fiber membrane bioreactor in biotechnology and tissue engineering is rapidly expanding. However there are some drawbacks of hollow fiber membrane for cell culture applications such as protein or small molecules fouling rate which lead to mass transport limitations. There arises a need for a bioreactor where cells can be grown efficiently with well define mass transport. This work focused on PES hollow fiber membrane surface coating with calcium alginate hydrogel is to overcome membrane bioreactor limitations mentioned above. Calcium alginate is well defined and studied for a long time since its excellent biocompatibility. We consider here a hydrophobic hollow fiber membrane coating with natural hydrogel method. Alginate coat was made by calcium ions diffused from membrane lumen to extracapillary space through membrane pores. Uniformly coated hydrogel was obtained by passive diffusion which was a gentle and precise fabrication method. This fabrication method is novel, fully controllable with simple and easy to manipulate fabricating conditions. Different cross-linker solvents and reaction time were investigated. The developed composite hollow fiber membrane was characterized by TEM for morphology. Hydrophilic test was conducted with water contact angle and hydraulic permeability. In addition, optical microscope was used for identifying the calcium alginate thickness. Our results imply calcium alginate coated hollow fiber membrane can be used as a membrane bioreactor with three dimensional scaffold or ultrafiltration system. 82 4.2 Introduction Hollow fiber membrane bioreactors (HFMB) have been widely used for cell culture in the biopharmaceutical and tissue engineering fields as well as for biomass filtration in water treatment units since 1972 [1, 2]. A typical hollow fiber membrane set-up consists of membranes arranged in a bundle and assembled within a module. This configuration provides a high surface area over volume ratio and therefore, has applications in continuous-perfusion culture systems. The set-up has been developed for use in mammalian cell culture [3, 4], microbial culture for ethanol production [5], mAb production [6–8], and enzymatic reactions [9, 10]. HFMB is also highly used for the purification of drinking water [11], in industry and municipal water treatment units [12], and dairy food processing [13]. The HFMB set-up has several advantages over static culture set-ups such as multi-wall plate or T-flask, including the ability to achieve high cell density. Static culture is a type of batch reactor in which nutrients, oxygen concentration, pH, and the accumulation of toxic metabolites within the flask over time follow a gradient structure [14]. In this method, there is a risk of contamination when the medium is being changed. Therefore, HFMB has been adopted in cell culture to provide or remove nutrients, growth factors, and metabolites through the porous wall of the membrane in a continuous manner [15]. Despite these significant advantages of HFMB, there are some limitations with respect to its mass transport ability [16]. In this method, cells are suspended in the extracapillary space and nutrients are provided through the membrane lumen. Nutrients diffuse through 83 the membrane pore driven by concentration differences, while the metabolites diffuse from the extracapillary side into the lumen. However, cells that are present at a large distance from the membrane have limited access to nutrients and oxygen, compared to cells that are present close to the membrane pore. In addition, these biological substances (nutrients and cytokines) tend to attract cells near the surface of the membrane, causing the cells to adhere on the surface. This results in differences in the physiological and biological states of cells at different distances from the membrane pores. Moreover, mass transport across the membrane is inhibited by the dense layer of cells that grow near the outer surface of the hollow fiber membrane. This increases the necrosis of cells within the HFMB. Therefore, HFMB is suitable only for bulk cell production within short periods (approximately 7 days) [17]. In addition, various components in water, such as natural organic matter (NOM) and proteins, cause fouling of the membrane [18, 19]. This decreases the flux and renders the process energy-intensive [20, 21]. Some recent studies have focused on improving the usability of HFMB in cell culture applications. To this end, three-dimensional tissue engineered constructs with scaffolds were proposed. Hollow fiber membranes have a capillary-like structure; on top of the membrane surface, a hydrophilic polymer was mixed with the cells for better inoculation in the extracapillary space. For example, Ye et al (2007) studied fibroblast cells by mixing them with collagen and Cytodex 1 microcarriers and injecting them into the extracapillary space of a cellulose acetate hollow fiber membrane. This inoculation treatment to generate a composite 3D scaffold resulted in a high cell density and 84 consistent lactate production. Ellis and Chaudhuri (2007) [22] developed a poly(lactic-coglycolide) (PLGA) hollow fiber membrane that can be used as clinical-scale bone constructs. This bioreactor enhances regeneration of bone tissues and provides an environment similar to the human circulatory system. Antwiler (2008) [23] demonstrated that the surface of a hollow fiber membrane may be treated with platelet lysate (PL), plasma, and fibronectin (FN), which are cell adhesion molecules, simply by dipping the membrane into the solution. Balmert et al. (2011) [24] evaluated the various circuit configurations applicable to bioreactors for measuring the volume shift of inner and outer fluids. The technical challenges involved in the process were resolved by designing complex tube circuits for better operating conditions. This configuration prevents shock due to changes in pressure and spikes in nutrient concentrations. Salzman (1999) [25] developed an immobilized enzyme reactor (by analytical modeling and numerical simulations) to obtain high conversions with intermediate flow rates and short operation times. Ye et al. (2006) [26] demonstrated nutrient transport in hollow fiber membrane bioreactors for 3D bone tissue. Efforts are being made to construct a bioreactor with improved membrane filtration and mass transport abilities. Polypropylene (PP), polyvinylidene fluoride (PVDF), polytetrafluoroethylene (PTFE), polysulfone (PS), and polyethersulfone (PES) materials are commonly used as separation layers because of their strong mechanical and chemical strengths. However, these materials are inherently hydrophobic. Such membranes were made more hydrophilic by using methods such as treatment with plasma [27, 28], grafting, 85 or coating [28–33], and blend modification [34, 35]. When pre-treated with ultra-violet rays, plasma, or chemical reagents, functional monomers grow on the membrane surface by initiating the active site [32, 36]. Although the membrane surface can be modified to be more hydrophilic, surface treatments are energy-intensive and require long modification times. Moreover, there is a high risk of un-polymerized monomer being left behind on the membrane, which leads to increased process costs [37]. The calcium alginate-coated PES hollow fiber membrane might be more cell-friendly, and have enhanced water permeability and mass transport abilities. Overall threedimension cell culture scaffolds provide high cell viability and targeted cell functionally [38, 39]. Sodium alginate is a natural polymer extracted from brown algae. It is a polysaccharide with (1-4)-linked β-D-mannuronate (M) and α-L-guluronic (G) residues. The ratio and sequence of M and G can be varied by treatment with chemical reagents during the extraction process [40, 41]. The alginate gel is cross-linked by divalent cations such as Ca2+, Ba2+, and Sr2+, which link the G residues [42]. Calcium alginate is a wellknown agent that is used for cell encapsulation, a process that involves dropping the alginate along with the cells (or drugs) into a calcium chloride bath. Within a specific cross-linking time, the cells or drugs are embedded in the calcium alginate matrix and are ready for further applications [43]. In addition, because the hydrogel is able to hold large volumes of water, it has been used as extracellular matrix in wound healing and tissue engineering [44]. It has low immunogenicity and toxicity, and an easy and simple crosslinking process; therefore, it has applications in the field of biochemical engineering. 86 In this study, we introduced alginate polymer onto a polyethersulfone (PES) hollow fiber membrane by cross-linking calcium ions diffusing through the membrane pores to improve membrane hydrophilicity. The degree of hydrophobicity was studied by evaluating the water contact angle. With this simple and gentle fabrication method, we achieved precisely controlled polymerization; moreover, the membrane was found to be biocompatible. The water permeability of this composite membrane was investigated by applying a positive pressure on the shell side of the membrane to drive water molecules into the membrane lumen. A small solute (dimethyl sulfoxide [DMSO]) was used to assess mass transfer limits for uncoated and coated membranes. DMSO is a common solvent used in chemical engineering. It is also used as a cryoprotectant to preserve living cells [44]. Coated membranes were analyzed by light and transmission electron microscopy (TEM). To the best of our knowledge, there is no other report that describes the coating of calcium alginate coated on a hollow fiber membrane using a simple crosslinker diffusion method. 4.3 Materials and Methods 4.3.1 Materials Sodium alginate (W201502), antipyrine ® (A5882), and DMSO (D4540) were purchased from Sigma-Aldrich, St. Louis, MO, USA. Calcium chloride (C77-500) and ethanol (BP2818) were obtained from VWR, Mississauga, ON, Canada. Hydrophilic 87 polyethersulfone (PES) hollow fiber membranes (Tetronic, 0.8mm i.d., 1.2 mm o.d., 150 kDa MWCO) was kindly donated by Hydranautics Inc., Japan. All tested solutions and reagents were prepared using ultra-pure water (18.2 MΩ-cm) obtained from a Diamond NANOpure water purification unit (Barnstead International, Dubuque, IA, USA), filtered just prior using by 0.2 μm pore size of Acrodisc® 25 mm syringe filter membranes (Pall Life Science, QC, Canada). 4.3.2 A Methods B C D Figure 1 The method for fabricating calcium alginate coating on ultrafiltration hollow fiber membrane as a substrate. A) cross section view of hollow fiber membrane lumen filled with water or 50 v/v% ethanol for pre-wetting step, B) after removal of water or 50 v/v% ethanol, introduce of sodium alginate solution in shell side at the mean while calcium chloride solution enter the membrane lumen, C) cross-linking of alginate radially outward diffusion of calcium ions 88 Sodium alginate solution (1 wt %) was prepared in ultrapure water. 0.1 M of calcium chloride solution was prepared in two solvents; one is water and the other is 50 v/v % ethanol. The solution was allowed to stand for 24 hours at 4 °C before use in order to evenly disperse the sodium alginate and to remove any entrapped air bubbles. Figure 1 shows some of steps involved in the coating process. The lumen of the hollow fiber membrane was filled with the base solvent (i.e. water or ethanol) for pre-wetting inner lumen of membrane presented in Figure 1A. This step could help better access of the calcium chloride solution into the hydrophobic (PES membrane) membrane pores. After removing base solvent from membrane lumen, the external and internal surface was patted-dry and placed in membrane module for coating process. This was done to prevent irregular cross-linking of alginate. The lumen was then filled with calcium chloride solution meanwhile shell side was filled with sodium alginate solution for the required duration in a given experiment (Figure 1B). Calcium ions passively diffused towards the alginate solution and ionically cross-linked alginates to be calcium alginate gel on the PES hollow fiber membrane (Figure 1C). After the given cross-linking duration, unreacted sodium alginate solution was rinsed using water (Figure 1D). Alginate coated hollow fiber membrane for tested by TEM were fixed with 2% Glutaraldehyde (2% v/v) in 0.1M sodium cacodylate buffer pH 7.4. The sample were then rinsed two times in buffer and post-fixed in 1 % osmium tetroxide in 0.1 M sodium cacodylate buffer for 1 hour. The samples were then dehydrated using graded ethanol series (i.e. 50%, 70%, 70%, 95% and 100%). The final dehydration for TEM samples was 89 done in 100 % propylene glycol (PPG). Infiltration with Spurr’s resin was carried out using graded series (i.e. PPG:Spurr’s resin ratios of 2:1, 1:1, 1:2, and lastly 100 % Spurr’s resin) with rotation of the samples in between solution changes. The samples were transferred to embedding molds with were then filled with fresh 100 % Spurr’s resin and polymerized overnight in a 60°C oven. Thin sections were cut using a Leica UCT Ultramicrotome and picked up onto Cu grids. The sections were post-stained with uranyl acetate and lead citrate and then viewed in a JEOL JEM 1200 EX TEMSCAN transmission electron microscope (JEOL, Peabody, MA, USA) operated at an accelerating voltage of 80kV. Figure 2 Experimental set-up for membrane characterization on hydraulic permeability at constant pressure, single PES hollow fiber membrane with effective length 30 cm, membrane module with effective inner volume 23.57 mL, and data acquisition (pressure and mass per time) with Logger pro. 90 Figure 2 shows the set-up used for the ultrafiltration experiment. The single PES hollow fiber was cut in 30 cm and fixed in the membrane module which was immersed within tap water for 30 min, for both coated and uncoated membranes. The content of feed tank was kept under pressure using compressed air. The pressure was applied in the feed tank as well as shell side of membrane module kept constant over the experiment. The permeate was collected by balance in every minute and was used to determine the permeate flux. Gas pressure sensor (Vernier software & Technology, Mississauga, Canada) and balance were connected to the computer for data acquisition using Lab Pro and Logger Pro (Vernier software & Technology, Mississauga, Canada). Figure 3 Experimental set-up for DMSO (300 g/L) diffusion experiment, PES ultrafiltration hollow fiber membrane with 30 cm effective length, Extracapillary space effective volume 5.89 mL, the pressure data collected and analyzed in Logger Pro (flow rate: 0.00866ml/min, pressure 101 kPa ) 91 Figure 3 shows the set-up used for the test solute and drug diffusivity experiment. The single PES hollow fiber placed in the membrane module. The feeder solutions (DMSO) were placed outside the hollow fibers (i.e. also referred to as extra-capillary space) while the receiver solution pumped through the membrane lumen with lowest flow rate (0.00866 ml/min) by two peristaltic pumps (Ismatec®, Cole-Parmer Instrument Company, Vernon Hills, IL, USA). Inner flow solution was recycled back to the feed tank which was kept well mixed. Pressure data was monitored by pressure sensor and inner solution sample was collected every 5min for UV absorbance. DMSO and antipyrine was analysed by NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) 4.4 Results and Discussions Figure 4 Magnified side view images of the uncoated/coated PES hollow fiber membrane. Images were obtained by light microscopy with a magnification factor of 25. Coating conditions: 1 wt% Alginate concentration, 0.1 M calcium chloride concentration in 50 v/v% ethanol, and 40 min cross-linking time 92 Figure 4 shows one example of fabrication of composite membrane carried out with ultrafiltration PES hollow fiber membrane using calcium alginate hydrogel. The experiment was carried out by 1 wt% of sodium alginate with cross-linked for 40 min by 0.1 M of calcium alginate solution in 50 v/v% EtOH. Top of the figure had uncoated PES hollow fiber membrane side view and the bottom sample was calcium alginate coated PES hollow fiber membrane. Natural hydrogels, such as alginate, collagen, agarose, and chitosan, have lower mechanical strength than synthetic hydrogels [45]. One of the features of hydrogel is environmental responsiveness under salt, pH, and temperature [46]. In this experiment, not only the coating process but also the tests with solutions that had salt presented for reversible collapse and swell of hydrogel were done. Once calcium alginate coated membrane was immersed into the 1M of ammonium sulphate solution, the hydrogel was collapsed. However, the collapsed hydrogel was not re-hydrated irreversibly, after water applied on dehydrated sample. As we expected, salt took the water body from the calcium alginate structure. At the mean time all the connections between the polymers broke. Under microscopy, the damaged hydrogel seemd like flakes on the membrane surface, which is not shown in figure. 93 Figure 5 Fabrication of calcium alginate coated hollow fiber membrane controllability test. Fabrication conditions: 1wt% alginate concentration, 0.1M of calcium chloride concentration, varied cross-linking time 0, 5, 10, 15, 20, and 40 min. The calcium alginate coating process was systemically controllable using different crosslinking times shown in Figure 5. This experiment was accommodated only with calcium chloride in 50 v/v % ethanol solution because calcium alginate solution in water based was not able to be verified for the hydrogel thickness below the level (25 times zoom) of light microscopes. The six of membranes were aligned side by side; the uncoated membrane (A), the once coated for 5 min (B), 10 min (C), 15 min (D), 20 min (E), and 40 min (F) with 0.1 M of calcium chloride cross-linker in 50 v/v % of ethanol. All the samples were cross-linked and put in storage in 0.1 M of calcium chloride solution for preventing deformation of hydrogel. Under the light microscope, samples were took snapshots for measuring the thickness using (Image J) software, which are presented in Table 1. The correlation between cross-linking time and thickness of hydrogel well fitted 94 first order (0-20 min). As a result of this experiment, the controllability of the amount of hydrogel deposit on membrane surface is high enough for various applications. Table 1. PES hollow fiber membrane coated with calcium alginate; 0.1 M calcium chloride solution was prepared in 50 v/v% ethanol, 1 wt % sodium alginate, in different cross-linking time Time Outer Diameter (n=4) 0 min 1.3 mm 5 min 1.53 ± 0.025 mm 10 min 1.74 ± 0.015 mm 15 min 1.89 ± 0.018 mm 20 min 2.34 ± 0.02 mm 40 min 2.99 mm Figure 6 Hollow fiber membrane coating with calcium alginate hydrogel A) Dew: calcium chloride solution pass through membrane pores B) 5 min cross-linking process C) 15 min cross-linking time. Fabrication condition: sodium alginate 1 wt%, calcium chloride 0.1 M in water, PES hollow fiber membrane 150 kDa MWCO. 95 Figure 6 shows another method for coating technique which the calcium chloride solution fully filled up under pressure in the membrane pores (Figure 6A). At t = 0, calcium ions are ready to cross-link alginate while the passive diffusion method requires time for calcium ions travel from inner membrane pore entrance to outer pore exit to meet alginate molecules. The difference is only where the calcium ion starts polymerization, the rest of fabricate conditions are the constant: diffusion coefficient of calcium chloride (in water 10-9m2/s) [47], tortuosity of pores, porosity of the membrane, concentration differences, and temperature. Because the calcium ions are ready to cross-linking, the calcium alginate gel are much thicker (Figure 6B). According to Cuadro et al. 2012 the estimated gelation of calcium alginate time was 40 sec for 2% of alginate and 0.5 % calcium chloride with 200 μm radius cylinder [48]. Calcium alginate polymerization is a rapid process if cross-linker availability is high enough. For this reason prefilled calcium chloride method showed more calcium alginate cross-linked. However, the roughness can be another point drawing consideration for various applications. Two samples in Figure 6 B and C showed different surface contours. The ultrafiltration hollow fiber membrane is in asymmetric conformation due to the extrusion manufacturing method, in which the pores presented unevenly on the hollow fiber wall compared to microfiltration membrane. The rough surface is a good indication of rapid calcium chloride polymerizing alginate from uneven location whereas passive diffusion process lead to controlled slow polymerization from membrane pores. This mass transport may counter the diffusion of alginate solution into membrane pores and slow calcium ions contacting, which generate calcium alginate gel morphologies. From this experiment, the application requires rapid 96 and thin layer of calcium alginate coating on the membrane surface for tissue engineering or immobilize cells in short period time at initial seeding step and it has more merits than passive method. Figure 7 TEM image obtained with the hollow fiber and composite membrane fabricated with 1 wt% sodium alginate and 0.1 M calcium chloride on PES ultrafiltration membrane, with a cross-linking time of 30 min. (A) control sample: uncoated membrane,25000 X magnification (B) 1 wt % of sodium alginate and 0.1 M calcium alginate in water, 5000 X magnification (C) bigger magnification of sample B, 20000X (D) 0.1M of calcium chloride solution in 50 v/v EtOH %, 20000 X magnification The calcium alginate coating morphology was observed by TEM analysis as shown in Figure 7. The samples were prepared three cases; one is uncoated membrane (A), second 97 is cross-linker solution dissolved in water (B and C), third one is cross-linker solution dissolved in 50 v/v% ethanol. Only the solvents for cross-linking agent were varied; concentrations of alginate, calcium chloride, and cross-linking time were kept constant. The control (uncoated PES hollow fiber membrane cross section) specimen is presented in Figure 7A. According to the degree of shade, there are three sections. Resin, top layer of membrane extracapillary side, and inner membrane were well indicated as expected. Figure 7 B and C are the same piece with different magnifications. The coated hydrogel thickness presented thinner than fresh piece because the preparation processes involved non-polar solvent caused dehydration of calcium alginate gel. Under higher magnification, hydrogel pore size and distribution is different in distance. The calcium alginate gel pore size has been gradually decreased toward to the edge of the outer alginate gel. Whereas ethanol based cross-linking cases (D) showed opposite trend. There are two possibilities to consider. One is calcium ion diffusivity in different solvent interactions with alginate base solution. Another point is ethanol wets membrane inner and pores faster than water so this enhances the calcium ions travel to alginate solution faster. The second possibility is alginate precipitating by solvent. Hassan reported much work about cross-linked alginate hydrogel where the forms go through the sol-gel state [49-51]. For better explanation of trend of calcium alginate formation, the compatibility of alginate solution and ethanol was investigated in Figure 8. 98 Figure 8 Calcium alginate polymerization study with different calcium chloride solvents (A: alginate + water based calcium chloride, B: alginate + 50v/v% ethanol based calcium chloride, C: alginate + 100 % ethanol, D: ethanol + alginate, E: sample D vertical pose), 1 wt% sodium alginate of 1mL, 0.1 M calcium chloride 300 μL Figure 8 illustrated four different cases of alginate and calcium chloride solutions. Concentration of alginate solution with 1ml mixed with calcium alginate solution 0.1M in water based (A), 50 v/v% ethanol (B). In comparison of two samples A and B, the mixture of alginate and calcium chloride well mixed in A, while two phases separate right after two liquid meet. Furthermore 100% of ethanol was injected in alginate solution (C) and rapidly alginate solution had chunk of sol with many bubbles. It suggested that ethanol induced the alginate into dehydrated state and precipitated first and after the solution was well mixed, aggregated alginate disappeared. Alginate aggregation was carried out in large volume of ethanol with small amount of alginate solution (D and E). This transition of alginate was well supported for Figure 7. From simple step of calcium alginate gel made of water based cross-linker case in Figure 7 C, this is a simple passive diffusion through the membrane pores and cross-linked alginate G residues. Moreover ethanol based cross-linker was one more step involved, first ethanol diffuse radically toward to shell side of alginate solution. Precipitate or aggregate alginate near the 99 membrane surface then calcium ions tides each alginate G residues. This is the reason why ethanol based calcium alginate had denser pores placed near the membrane surface. Figure 9 Fabrication of calcium alginate coated on PES hollow fiber membrane (Alginate concentration 1wt%, calcium chloride concentration 0.1M and 1 min crosslinking time) optical microscope 25 times. Figure 9 presented the fabrication of calcium alginate composite membrane that was carried out with 1 wt% of alginate, 0.1 M of calcium chloride solution in water. The cross-linking was done for 1min. Since the short exposure time to cross-linking agent, the calcium alginate hydrogel generated was too thin to measure under optical microscope. However, top sample is corresponding to uncoated membrane with rough/scratched skin while bottom sample has clean and smooth surface. Even though calcium alginate coating layer is not able to observe, light reflections of the samples are different. For more membrane characterization in terms of hydrophilized surface assessment, contact angle analysis was carried out. 100 Figure 10 Water contact angle analysis with PES hollow fiber membrane (left), and calcium alginate coated membrane (right). Picture obtained by digital camera. Contact angle was tested to identify the hydrophobicity of coated and uncoated membrane surfaces. The same samples were used from Figure 10. Contact angle can provide information about the interfacial forces of water and membrane surfaces shown in Figure 8. Left side of sample is uncoated PES membrane that is relatively hydrophobic than right side of sample. Coated membrane is friendlier to water which pulls the water toward to the membrane surface, which is indicated by the water contour changes. This wetting effect is an indication of the membrane surface hydrophilicity improvement. 101 50 45 40 35 30 25 20 15 10 5 0 Uncoated membrane coated membrane 2 4 6 8 10121416182022242628 Filtration Time (min) Figure 11 Measurement of water flux and hydraulic permeability of uncoated/coated PES hollow fiber membrane, operating pressure at 109.45 kPa (uncoated membrane) and 110.56 kPa (coated membrane) with constant (one data set). Figure 11 shows the water flux and hydraulic permeability of the uncoated and coated hollow fiber membranes. The membrane was prepared in the same condition with Figure 9, and 10 with 1 min cross-linking time of 1wt% alginate concentration and 0.1 M of calcium chloride. The experimental set up is explained in Figure 2. The water flux was calculated by 𝑊𝑎𝑡𝑒𝑟 𝑓𝑙𝑢𝑥 = Permeated water volume Membrane area × Filtration time 102 as shown in Figure 11. Since the pressure applied by compressed air was not exactly same value in two cases, hydraulic permeability can be a factor to normalize to overlook the membrane filtration behaviour with consideration of pressure term. The hydraulic permeability was obtained by 𝑃𝑒𝑟𝑚𝑒𝑎𝑏𝑖𝑙𝑖𝑡𝑦 = Flux × Viscosity of water Transmembrane pressure [50], where the viscosity of water is 0.000145 pa.s. The hydraulic permeability also had the trend to decrease over the filtration volume. Coated membrane has similar water flux behaviour. There are two possibilities for this phenomenon. One is fouling behaviour over filtration time. The second one is coated hydrogel was in fully hydrated state prior the filtration process and then the pressure applied from outside to inside made hydrogel collapse, which increased the resistance of permeate flux. The water permeability of coated membrane had higher than uncoated membrane because calcium alginate was not working as a barrier, on the contrary it helps penetration of water using hydrophilic characteristic of the hydrogel. Sawada et al. 2012 reported PES hollow fiber membrane was grafted on acrylamide incubating silver nanoparticles. It shows hydrophilicity improved by contact angle measure value however the water permeability was decreased by grafting layer [53]. It is suggested that even one more layer of hydrophilic hydrogel presented on the membrane surface, the hydraulic permeability does not change membrane performance. 103 Figure 12 Effect of coating hollow fiber membrane on DMSO mass transport with Fig 3 experimental set up (one data set). Figure 12 shows the effect of coating hollow fiber membrane by solute diffusion from shell side of membrane to inner lumen. It was obtained with 300 g/L of DMSO feed solution which slowly diffused through the membrane pores into the circulated water in inner lumen during 200 minutes the circulation ran with 0.00866ml/min flow rate. Samples were collected from the permeate reservoir by 10 uL for monitoring DMSO concentration. The increase of DMSO concentration in membrane lumen is good indicator for small solute transport through membrane pores. At the beginning of diffusion experiment (0-100 min), the DMSO diffusion rate was higher because the concentration difference between two sides (inner and outer) of membrane was high. In this experiment, pressure is atmosphere and it means the concentration difference of shell 104 and lumen side is mass transport driving forces. After 100 min of diffusion run, the flux of DMSO decreased and reached a plateau. This experiment suggested the calcium alginate coated membrane had similar mass transport behaviour. 4.5 Conclusions Calcium alginate hydrogel was successfully coated on PES hollow fiber membrane by passive diffusion of cross-linking agent through membrane pores. This coating technique is gentle, precisely controllable on hydrogel thickness, as well as simple in experimental set-up. Effect of cross-linking agent solvent was studied on thickness and morphology of calcium alginate by TEM analysis. Calcium alginate coated membrane was characterized by water contact angle as well as optical microscope. Since calcium alginate has been used in tissue engineering with good biocompatibility, this technique will offer many advantages: tissue engineering application for three dimensional MBR with similar hydraulic permeability, material diffusion rate, and better cell adhesion effect due to the hydrophilic coated on membrane. Calcium alginate coating made water favorable for easy permeation and this characteristic helps the water purification process, too. However, there are lots of room to improve this technique. Each proteins or simulated cell culture medium can be tested with coated membrane for tissue engineering aspect, and several coated membranes put as a bundle for tests of the water filtration performance since uncoated and coated membrane indicated similar hydraulic permeability. 105 4.6 Reference [1] A.S. Michaels, C.R. Robertson, S.N. Cohen, Microbiological methods using hollow fiber membrane reactor, (1984) US4440853 [2] R.A. Knazek, P.M. Gullino, P.O. Kohler, R.L. Dedrick, Cell Culture on Artificial Capillaries: An Approach to Tissue Growth in vitro, Science. 178 (1972) 65–67. [3] D.Q. Wu, G.L. Zhang, C. Shen, Q. Zhao, H. Li, Q. Meng, Evaluation of diffusion in gel entrapment cell culture within hollow fibers., World J. Gastroenterol. 11 (2005) 1599–604. [4] H. Gloeckner, H.D. Lemke, New miniaturized hollow-fiber bioreactor for in vivo like cell culture, cell expansion, and production of cell-derived products, Biotechnol. Prog. 17 (2001) 828–31. [5] D. Inloes, D. Taylor, Ethanol production by Saccharomyces cerevisiae immobilized in hollow-fiber membrane bioreactors, Appl. Environ. Microbiol. 46 (1983) 264–278. [6] P.J. Yazaki, L. Shively, C. Clark, C.W. Cheung, W. Le, B. Szpikowska, J.E. Shively, A.A. Raubitschek, A.M. Wu, Mammalian expression and hollow fiber bioreactor production of recombinant anti-CEA diabody and minibody for clinical applications, J. Immunol. Methods. 253 (2001) 195–208. [7] L.R. Jackson, L.J. Trudel, J.G. Fox, N.S. Lipman, Evaluation of hollow fiber bioreactors as an alternative to murine ascites production for small scale monoclonal antibody production, J. Immunol. Methods. 189 (1996) 217–31. [8] D. Lowrey, S. Murphy, R.A. Goffe, A comparison of monoclonal antibody productivity in different hollow fiber bioreactors, J. Biotechnol. 36 (1994) 35–8. [9] P. Ye, Z.K. Xu, A.F. Che, J. Wu, P. Seta, Chitosan-tethered poly(acrylonitrile-comaleic acid) hollow fiber membrane for lipase immobilization, Biomaterials. 26 (2005) 6394–403. [10] H. Kawakita, K. Sugita, K. Saito, M. Tamada, T. Sugo, H. Kawamoto, Production of cycloisomaltooligosaccharides from dextran using enzyme immobilized in multilayers onto porous membranes, Biotechnol. Prog. 18 (2002) 465–9. 106 [11] S. Nakatsuka, I. Nakate, T. Miyano, Drinking water treatment by using ultrafiltration hollow fiber membranes, Desalination. 106 (1996) 55–61. [12] C. Lubello, R. Gori, Membrane bio-reactor for textile wastewater treatment plant upgrading, water science and technology, J. Int. Assoc. Water Pollut. Res. 52 (2005) 91–98. [13] Y. Pouliot, Membrane processes in dairy technology – from a simple idea to worldwide panacea, Int. Dairy J. 18 (2008) 735–740. [14] L.K. Nielsen, Bioreactors for hematopoietic cell culture, Annu. Rev. Biomed. Eng. 1 (1999) 129–52. [15] U. Marx, H. Matthes, A. Nagel, R. Baehr, Application of a hollow fiber membrane cell culture system in medicine, Am. Biotechnol. Lab. (1993) 26. [16] A. Ouyang, S. T. Yang, A two-stage perfusion fibrous bed bioreactor system for mass production of embryonic stem cells., Expert Opin. Biol. Ther. 8 (2008) 895– 909. [17] H. Ye, Z. Xia, D.J.P. Ferguson, J.T. Triffitt, Z. Cui, Studies on the use of hollow fibre membrane bioreactors for tissue generation by using rat bone marrow fibroblastic cells and a composite scaffold., J. Mater. Sci. Mater. Med. 18 (2007) 641–8. [18] H. Evenblij, J.H. van der Graaf, Occurrence of EPS in activated sludge from a membrane bioreactor treating municipal wastewater, Water Sci. Technol. 50 (2004) 293–300. [19] N. Park, B. Kwon, I.S. Kim, J.W. Cho, Biofouling potential of various NF membranes with respect to bacteria and their soluble microbial products (SMP): characterizations, flux decline and transport parameters, J. Membr. Sci. 258 (2005) 43–54. [20] A. Bjørkøy, L. Fiksdal, Characterization of biofouling on hollow fiber membranes using confocal laser scanning microcscopy and image analysis, Desalination. 245 (2009) 474–484. [21] I. Chang, P. Le Clech, B. Jefferson, S. Judd, Membrane Fouling in Membrane Bioreactors for Wastewater Treatment, (2002) 1018–1029. [22] M. Ellis, J. Chaudhuri, Poly (lactic-co-glycolic acid) hollow fibre membranes for use as a tissue engineering scaffold, Biotechnol. Bioeng. 96 (2007) 177–187. 107 [23] G.D. Antwiler, Improved bioreactor surfaces, (2008) WO2008124229 A2 [24] S.C. Balmert, D. McKeel, F. Triolo, B. Gridelli, K. Zeilinger, R. Bornemann,J.C. Gerlach, Perfusion circuit concepts for hollow-fiber bioreactors used as in vitro cell production systems or ex vivo bioartificial organs, Int J Artif Organs. 34 (2011) 410–421. [25] G. Salzman, R. Tadmor, S. Guzy, S. Sideman, N. Lotan, Hollow fiber enzymic reactors for a two substrate process: analytical modeling and numerical simulations, Chem. Eng. Process. Process Intensif. 38 (1999) 289–299. [26] H. Ye, D.B. Das, J.T. Triffitt, Z. Cui, Modelling nutrient transport in hollow fibre membrane bioreactors for growing three-dimensional bone tissue, J. Memb. Sci. 272 (2006) 169–178. [27] D.S. Wavhal, E.R. Fisher, Membrane surface modification by plasma-induced Polymerization of Acrylamide for Improved Surface Properties and Reduced Protein Fouling, Langmuir. 19 (2003) 79–85. [28] H.Y. Yu, H. Lei, Z.K. Xu, Enhencement of the flux for polypropylene hollow fiber membrane in a submerged membrane-bioreactor by surface modification, J. Environ. Sci. (China). 18 (2006) 1050–1055. [29] A. Bhattacharya, B.N. Misra, Grafting: a versatile means to modify polymersTechniques, factors and applications, Prog. Polym. Sci. 29 (2004) 767– 814. [30] W. Sun, J. Liu, H. Chu, B. Dong, Pretreatment and Membrane Hydrophilic Modification to Reduce Membrane Fouling, Membranes (Basel). 3 (2013) 226– 241. [31] Y.J. Shen, G.X. Wu, Y.B. Fan, H. Zhong, L.L.Wu, S.L. Zhang, X.H. Zhao, W.J. Zhang, Performances of biological aerated filter employing hollow fiber membrane segments of surface-improved poly (sulfone) as biofilm carriers, J. Environ. Sci. (China). 19 (2007) 811–7. [32] M. Taniguchi, G. Belfort, Low protein fouling synthetic membranes by UVassisted surface grafting modification: varying monomer type, J. Memb. Sci. 231 (2004) 147–157. 108 [33] W. Junfu, Z. Kongyin, W. Lei, Z. Huan, C. Li, Z. Zhengpu, PVDF Hollow fiber Membrane with High Flux and High Rejection Ratio Prepared by Irradiation Induced Grafting of PAA, Macromol. Symp. 297 (2010) 231–239. [34] Q.P. Pham, U. Sharma, A.G. Mikos, Electrospinning of polymeric nanofibers for tissue engineering applications: a review., Tissue Eng. 12 (2006) 1197–211. [35] C. Liu, R. Bai, Adsorptive removal of copper ions with highly porous chitosan/cellulose acetate blend hollow fiber membranes, J. Memb. Sci. 284 (2006) 313–322. [36] J. Pieracci, J. V Crivello, G. Belfort, Photochemical modification of 10 kDa polyethersulfone ultrafltration membranes for reduction of biofouling, J. Memb. Sci. 156 (1999) 223–240. [37] H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu, J. Hirvonen, Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam)., Biomaterials. 26 (2005) 3055–3064. [38] Y. Luo, G. Engelmayr, D.T. Auguste, S. Ferreira, J.M. Karp, R. Saigal, R. Langer, 3-dimensional scaffolds, in Principles of tissue engineering, 3rd edition, R. Lanza, R. Langer, J.P. Vacanti, Elsevier/Academic press, 2007, 359-373. [39] M. Lovett, K. Lee, A. Edwards, D.L. Kaplan, Vascularization strategies for tissue engineering., Tissue Eng. Part B. Rev. 15 (2009) 353–70. [40] G.T. Grant, E.R. Morris, D.A. Rees, P.J.C. Smith, D. Thom, Biological interactions between polysaccharides and divalent cations: The egg-box model, FBES Lett. 32 (1973) 195–198. [41] K.I. Draget, G. Skjåk-Braek, O. Smidsrød, Alginate based new materials, Int. J. Biol. Macromol. 21 (1997) 47–55. [42] O. Smidsrød, G. Skjåk-Braek, Alginate as immobilization matrix for cells, Trends Biotechnol. 8 (1990) 71–8. [43] Y.M. Kolambkar, K.M. Dupont, J.D. Boerckel, N. Huebsch, D.J. Mooney, D.W. Hutmacher, R.E. Guldberg, An alginate-based hybrid system for growth factor delivery in the functional repair of large bone defects, Biomaterials. 32 (2011) 65– 74. 109 [44] D. Pegg, Cryopreservation and freeze-drying protocols, in: J. Day, G. Stacey (Eds.), Methods in Molecular Biology (Humana Press), 2007: pp. 39–57. [45] K.Y. Lee, D.J. Mooney, Hydrogels for Tissue Engineering, Chem. Rev. 101 (2001) 1869–1880. [46] J. Kopecek, Hydrogel biomaterials: a smart future?, Biomaterials. 28 (2007) 5185– 5192. [47] A.C.F. Ribeiro, M.C.F. Barros, A.S.N. Teles, A.J.M. Valente, V.M.M. Lobo, A.J.F.N. Sobral, M.A. Esteso, Diffusion coefficients and electrical conductivities for calcium chloride aqueous solutions at 298.15K and 310.15K, Electrochim. Acta. 54 (2008) 192–196. [48] T.R. Cuadros, O. Skurtys, J.M. Aguilera, Mechanical properties of calcium alginate fibers produced with a microfluidic device, Carbohydr. Polym. 89 (2012) 1198–1206. [49] R. Hassan, A. Gobouri, I. Zaafarany, S.A. Kingdom, Kinetics and mechanism of Sol-Gel transformation between sodium alginate anionic polyelectrolyte and some alkaline earth metal ions with formation of coordination biopolymer ionotropic polymembrane hydrogels of capillary structure, ABB (2013) 47-56. [50] M. Mahmoud, R. Abdel-Kader, M. Hassanein, S. Saleh, S. Botros, Antipyrine clearance in comparison to conventional liver function tests in hepatitis C virus patients, Eur. J. Pharmacol. 569 (2007) 222–7. [51] R. Hassan, F.Tirkistani, I. Zaafarany, A. Fawzy, M. Khairy, S. Iqbal, Polymeric biomaterial hydrogels. I. Behavior of some ionotropic cross-linked metal-alginate hydrogels especially copper-alginate membranes in some organic solvents and buffer solutions, ABB 3 (2012) 845-854. [52] J. Kestin, M. Sokolov, W. A. Wakeham, Viscosity of liquid water in the range −8 °C to 150 °C, J. Phys. Chem. Ref. Data. 7 (1978) 941. [53] I. Sawada, R. Fachrul, T. Ito, Y. Ohmukai, T. Maruyama, H. Matsuyama, Development of a hydrophilic polymer membrane containing silver nanoparticles with both organic antifouling and antibacterial properties, J. Memb. Sci. 387-388 (2012) 1–6. 110 Chapter 5 Fabrication of alginate fibers using a microporous membrane based molding technique 4 4 This chapter is organized based on a manuscript written by Seung Mi Yoo and Raja Ghosh. This work has been submitted and under the minor revision process to Biochemical Engineering Journals (Manuscript #BEJ-D-14-00034) 111 5.1 Abstract Tubular structures made of alginate and similar hydrogels have been extensively investigated for drug delivery, tissue engineering and other biomedical applications. Alginate fibers are usually fabricated using complicated techniques such as coaxial flow extrusion or electro-spinning. This paper discusses the fabrication of hollow and solid alginate fibers using a simple membrane based molding technique. A hollow-fiber microfiltration membrane served both as the mold as well as reservoir for the crosslinking agent. The pores of the membrane were first filled with the cross-linker (i.e. calcium chloride) solution, after which the lumen was filled with sodium alginate solution. The calcium ions diffused from the membrane pores into the lumen, cross-linking the alginate in a radially inward direction. Solid alginate fibers were obtained by crosslinking all the alginate within the lumen, while hollow fibers were fabricated by pushing out un-cross-linked alginate from the central core using calcium chloride solution. The alginate fibers fabricated as described above were expelled from the membrane mold using water under pressure. These were then characterized by optical, scanning electron, and transmission electron microscopy. The most attractive features of this fabrication method are its simplicity and flexibility with regards to alginate concentration. The fibers obtained were straight with excellent definition and uniform thickness, and could be fabricated in a highly reproducible manner. Other complicated solid and hollow 3dimensional structures suitable of biomedical, and indeed other applications could also potentially be fabricated using similar membrane-based molding techniques. 112 5.2 Introduction Hydrogels can hold large amounts of water within their structures and are widely used for a range of pharmaceutical and biomedical applications [1,2]. Synthetic hydrogels such as cross-linked polyethylene glycol (PEG), poly (N-isopropylacrylamide) (PNIPAM) and poly-hydroxyethylmethacrylate (PHEMA) can be tailored to specific chain lengths and degree of cross-linking, and can also be functionalized to make them stimuli-responsive, an attribute extremely useful for controlled or sustained release of drugs [3]. However, un-polymerized monomer residues can be highly toxic, and their complete removal can prove to be challenging. Consequently, natural hydrogels continue to be widely used for drug delivery, cell culture and tissue engineering applications, where toxicity due to monomers and cross-linkers could be a major issue [4]. Natural hydrogels include biopolymers such as alginate, collagen, hyaluronic acid (HA), fibrin, agarose, chitosan, and combinations of two or more of these [5]. Alginate, which is obtained from brown algae is the most popular amongst these, having been widely used since the 1980s, due to its low immunogenicity and low toxicity, for cell encapsulation, drug delivery, woundhealing, and tissue engineering [6-8]. It is a polysaccharide with (1-4)-linked β-Dmannuronate (M) and α-L guluronic acid (G) residues, with the sequence of M and G residues varying, depending on the source. The gelation of alginate is induced by the presence of divalent cations such as Ba2+, Sr2+, and Ca2+ which interact with the G residues [6, 7]. Due to the involvement of the G residue in cross-linking, the G/M ratio affects the mechanical strength and biodegradability of alginate [7, 8]. The formation of calcium alginate gel has been described based on an “egg-box model” [8]. 113 Spherical calcium alginate beads have been widely used for cell encapsulation and drug delivery due to the simple fabrication method, i.e. drop-wise addition of sodium alginate solution into cross-linker (calcium chloride) solution [7]. The material to be encapsulated is typically pre-mixed with the sodium alginate solution prior to cross-linking. Some of the drawbacks linked with spherical alginate beads and their fabrication method are a) loss of cells or drug into the cross-linking solution, b) the so called “burst-effect” whereby there is an initial rapid release of drug from the beads to any recipient solution, and c) nutrient mass transport limitations at the centre of the beads which could lead to cell necrosis [9-12]. The drop-wise bead production methods also leads to size distributions, and this makes precise drug dosing difficult. A critical limiting factor for the spherical geometry is that the surface area to volume ratio is determined by the radius, thereby limiting flexibility in designing structures. Alternative geometries such as tubes, rods and cylinders have been shown to have some advantages in certain drug delivery and tissue engineering applications [13]. With the cylindrical form, dosage can be very precisely controlled based on length and diameter. Also, the surface area to volume ratio is decoupled from the radius and can be flexibly manipulated by altering both the radius and length of the structure. Moreover, in certain applications such as nerve and vascular tissue engineering, the fiber is the only suitable scaffold geometry. Electro-spinning, one of the well-established methods for fabricating fibers, has attracted much attention due the ease with which fine fibers with diameters ranging from 10 nm to 100 micrometers can be produced using both synthetic and natural hydrogel materials 114 [14-20]. However, this technique involves the application of high voltages, e.g. 10~15 kV[17], 20 kV [18], 13~15 kV [19], and 10~50 kV [20], which could potentially lead to cell mortality in cell immobilization and tissue engineering applications. During cellembedded fiber fabrication by electro-spinning, cell death was as high as 60 % [21]. Moreover, electro-spinning is not particularly suitable for fabrication of fibres with outer diameter greater than 100 micrometer [17, 19, 22]. Another major problem with electrospinning of alginate is that the method is restricted to the use of alginate solutions of lower than 2% concentration due to viscosity issues, and consequently fibers with low mechanical strengths are produced[22]. Extrusion based technique have also been widely used to produce alginate fibers. For instance, Li et al [11] reported a very simple technique for producing alginate cylinders by directly injecting sodium alginate into calcium chloride solution. Improved versions of this technique involving sophisticated multichannel nozzles, suitable for fabricating more complicated structures have been subsequently reported [23-25]. These techniques were suitable for producing 100~800 μm diameters fibres by manipulating variables such as reagent concentration and extrusion velocity, and by using sheath solutions [26]. The combination of extrusion and microfluidic techniques have been shown to enhance the consistency of fiber production [27-29]. However higher concentrate alginate solutions cannot be satisfactorily handled by microfluidic systems as their high viscosity makes it difficult to balance the central flow and sheath flows, which in turn leads to nonuniformity in fiber dimension. Moreover the high shear stress observed in such system 115 could be problematic in cell immobilization and tissue engineering applications. In an effort to reduce shear stress, a roller assisted microfluidic system was developed whereby alginate fibres produced within microfluidic channels were drawn out in a controlled manner [30]. Other techniques reported for fabrication of alginate fibers include 3D printing [31], laser-assisted printing [32] and the use of centrifugal force [33]. Boland et al. [34] have reviewed the use of 3D printing in tissue engineering including the fabrication of alginate structures. In this paper, we describe a molding technique, using which hollow and solid alginate fibers can be fabricated under gentle and sterile conditions. A hollow-fiber micro- or ultra-filtration membrane is used as the mold for fabricating these alginate fibers. The pores present on the hollow fiber membrane served as the reservoir for calcium chloride solution was used to cross-link alginate within the fiber lumen. The method is summarized in Figure 1. As shown in A, the pores of the hollow fiber were first filled with calcium chloride solution. The lumen of the hollow fiber was then filled with sodium alginate solution (B). The divalent calcium ions diffused from the pores into the lumen, cross-linking the alginate in a radially inward direction (C). For producing solid fibers, the alginate held within the lumen was allowed to fully cross-link before expelling it from the hollow-fiber membrane. For producing alginate hollow fibers, the crosslinking was allowed to take place till the desired wall thickness was attained followed by removal of uncross-linked alginate from the interior core using calcium chloride solution (D). This step also resulted in the curing of the inner surface of the alginate hollow fiber, 116 as well as delamination of alginate fiber from substrate membrane by dehydration which was then recovered by expulsion from the mold under pressure using water or calcium chloride solution (Figure 1 E). Alginate fibers of different sizes and geometries were fabricated using the above technique. Effects of variables such as alginate concentration, cross-linker concentration and cross-linking time on the fiber dimension were examined to demonstrate the controllability of the fabrication technique. The alginate fibers produced were tested using techniques such as scanning electron microscopy (SEM), tunneling electron microscopy (TEM), and light and fluorescence microscopy. The feasibility of using the molding techniques for tissue engineering and cell immobilization applications was demonstrated by fabricating alginate hollow fibers containing live human umbilical vein endothelial cells (HUVEC). 117 Figure 1 The method for fabricating calcium alginate hollow fibers using a porous hollow fiber membrane as a mold (A) cross sectional view of hollow fiber membrane with pores filled with calcium chloride solution, (B) sodium alginate solution being filled inside the lumen of the hollow fiber membrane, (C) cross-linking of alginate by radially inward diffusion of calcium ions, (D) removal of un-cross-linked alginate core with calcium chloride solution, and (E) expulsion of calcium alginate hollow fiber from mold. 5.3 Materials and Methods Sodium alginate (W201502), and FITC-dextran (46947; excitation 490 nm, emission 520 nm) were purchased from Sigma-Aldrich, St. Louis, MO, USA. Calcium chloride (C77500) was obtained from VWR, Mississauga, ON, Canada. Polypropylene hollow fiber microfiltration membranes (Accurel PP S6/2, 1.8 mm i.d., 2 mm o.d., 0.2 μm pore size; Accurel Q3/2, 0.6 mm i.d., 1 mm o.d., 0.2 micron pore size) were purchased from Membrana, Wuppertal, Germany. Hydrophilic polyethersulfone hollow fiber 118 ultrafiltration membranes (Tetronic, 0.8 mm i.d., 1.2 mm o.d., 150 kDa MWCO) was kindly donated by Hydranautics Inc., Japan. All test solutions and reagents were prepared using ultra-pure water (18.2 mΩ-cm) obtained from a Diamond Nanopure water purification unit (Barnstead International, Dubuque, IA, USA), vacuum-filtered just prior to use using 0.45 μm pore size cellulose acetate membrane (Nalgene Nunc International, Rochester, NY, USA). 5.3.1 Fabrication of calcium alginate hollow fibers Figure 2 Sequence of events in calcium alginate hollow fiber molding process (A) sweating of calcium chloride on the outer wall of the hollow fiber membrane indicating that the pores are filled, (B) removal of un-cross-linked sodium alginate core from the mold using calcium chloride solution, (C) expulsion of calcium alginate hollow fiber from mold using water, (D) alginate hollow fiber after recovery from mold. 119 Sodium alginate solutions of different concentration (e.g. 1, 2.5, 5 wt %) were prepared in ultrapure water. The solutions were allowed to stand for 24 hours at 4°C before use in order to evenly disperse the polymer and to remove any entrapped air bubbles. Calcium chloride (cross-linker) solutions of different concentrations (e.g. 0.1, 0.5, 1 M) were also prepared in ultrapure water. All the materials used for alginate fiber fabrication were sterilized and transferred to a sterile laminar flow cabinet. Figure 2 shows some of the steps involved in the fabrication process. The lumen of the hollow fiber membrane was first filled with calcium chloride under pressure to fill up the pores present on the membrane. Sweating on the outer surface of the membrane (see A) confirmed that the membrane pores were filled with cross-linker. The calcium chloride solution was emptied from the lumen, the external surface was patted-dry and air was passed through the lumen to remove excess calcium chloride solution from the inner surface. This was done to prevent irregular cross-linking of alginate during the fabrication process. The lumen was then filled with sodium alginate solution which was maintained inside for the required duration in a given experiment. For preparing alginate hollow fibers, uncross-linked material present in the core was pushed out using calcium chloride solution (see B). The fiber fabricated within the hollow fiber membrane was expelled using water under pressure (see C). The filling, rinsing and pressurising steps described above were carried out using sterile plastic syringes. 120 5.3.2 Microscopy Alginate fiber samples for testing by TEM and SEM were fixed with 2% Glutaraldehyde (2% v/v) in 0.1 M sodium cacodylate buffer pH 7.4. The samples were then rinsed two times in buffer and post-fixed in 1 % osmium tetroxide in 0.1 M sodium cacodylate buffer for 1 hour. The samples were then dehydrated using graded ethanol series (i.e. 50%, 70%, 70%, 95%, 95%, and 100%). The final dehydration for TEM samples was done in 100% propylene glycol (PPG). Infiltration with Spurr's resin was carried out using graded series (i.e. PPG:Spurr’s resin ratios of 2:1, 1:1, 1:2, and lastly100% Spurr's resin) with rotation of the samples in between solution changes. The samples were transferred to embedding moulds which were then filled with fresh 100% Spurr's resin and polymerized overnight in a 60 °C oven. Thin sections were cut using a Leica UCT Ultramicrotome and picked up onto Cu grids. The sections were post-stained with uranyl acetate and lead citrate and then viewed in a JEOL JEM 1200 EX TEMSCAN transmission electron microscope (JEOL, Peabody, MA, USA) operated at an accelerating voltage of 80kV. The samples for SEM were dehydrated in 100% ethanol and then critical point dried. These were then mounted onto SEM stubs, sputter-coated with gold and then viewed in a Tescan Vega II LSU scanning electron microscope (Tescan USA, PA) operated at 20kV. A florescence inverted microscope- Zeiss Axiovert 100M microscope was used for confocal laser scanning microscopy of alginate fibers. All light microscopy images were acquired with a Zeiss AxioCam HRc camera. AxioVision software was used for transferring and analyzing images. 121 5.3.3 Human umbilical vein endothelial cell culture Human umbilical vein endothelial cells or HUVEC (C-003-5C) were purchased from Gibco Life Technology (Burlington ON, Canada). HUVEC was cultured by supplementing low serum growth factors (EGM-2, CC-4176) in endothelial cell basal medium containing 1% v/v penicillin-streptomycin (P/S) (Cat #15070063). The cells were cultured at 37 °C in 5 % carbon dioxide and 100 % relative humidity. The media was changed every second day. Once the cells attained 100 percent confluence, cells were washed with 12 mL of 0.25% Trypsin/EDTA (Invitrogen, 12604021) for 3 min to detach the cells from the inner surface of the T-75 culture flask used for cell culture. The cell suspension thus obtained was transferred to a sterile 15 mL falcon tube and centrifuged at 14,000 rpm for 5 min. The cell pellet which had approximately 9 × 106 cells/mL was dispersed in sterile sodium alginate solution to fabricate cell-containing alginate fibers. 122 5.4 Results and Discussions Figure 3 Alginate fibers fabricated using hollow fiber membrane based molding technique: (A) Fibers made with 2.5% sodium alginate, 0.1 M calcium chloride solution as cross-linker using (from the top) Accurel Q3/2, Tetronic and Accurel PP S6/2 hollow fiber membrane molds respectively. The cross-linking times were 10, 30 and 60 seconds respectively for the small, medium and large diameter hollow fibers. (B)Solid and (C) hollow fibers fabricated with 5% sodium alginate, 1 M calcium chloride solution as cross-linker, using Accurel PP S6/2 membrane as mold. The cross-linking times were 600 seconds and 60 seconds respectively. Figure 3 shows some of the solid and hollow alginate fibers fabricated using different hollow fiber membranes as molds, using different reagents and cross-linking time. The hollow fibers shown in Figure 3A were made using 2.5% sodium alginate solution, and 0.1 M calcium chloride solution as cross-linker. The cross-linking times were 10, 30 and 60 seconds respectively for the small, medium and large diameter hollow fibers. The 123 small diameter hollow fiber (200 µm o.d., 50 µm i.d.) was molded using Accurel Q3/2 membrane, the medium diameter hollow fiber (600 µm o.d., 375 µm i.d.) using Tetronic membrane, and the large diameter hollow fiber (1.6 mm o.d., 0.6 mm i.d.) using Accurel PP S6/2 membrane. The solid and hollow fibers shown in Figures 3B and 3C respectively were fabricated using 5% sodium alginate with 1 M calcium chloride solution as crosslinker, using Accurel PP S6/2 membrane as mold. The only difference between the two was with regards to cross-linking time. While the solid fibers were obtained by a 600 second cross-linking process, the hollow fibers were obtained by 60 seconds of crosslinking. As expected, the outer diameter of the alginate fiber was dependent on the inner diameter of the hollow fiber membrane mold. Also, for a given mold, the inner diameter could be controlled through the cross-linking time and hollow or solid fibers, as required could be produced. In addition to the differences in dimension, these fibers also differed significantly in terms of their rigidity. The fibers produced using 2.5% sodium alginate solution was softer than those produced using 5% alginate. The fibers could be produced in a highly reproducible manner, simply by controlling the reagent concentrations and cross-linking time. The individual solid fibers shown in Figure 3B were separately fabricated and were found to have fairly consistent outer diameter of 1.35 ± 0.08 mm (n=8). Similarly, the hollow fibers shown in Figure 3C were separately fabricated and found to have consistent outer diameter of 1.42± 0.06 mm (n=8) and inner diameter of 0.65± 0.004 mm (n=8) mm. The slightly lower outer diameter of the solid fiber suggests that the greater extent of cross-linking caused some inward shrinkage of the structure. 124 Figure 4 Magnified side view (A) and cross-sectional view (B) images of the alginate hollow fiber shown in Figure 3 C. Images were obtained with the fiber immersed in water by light microscopy with a magnification factor of 25. Figure 4 shows magnified cross-section and side view images of the alginate hollow fiber shown in Figure 3 C. These images were obtained under immersed condition (in water) by light microscopy with a magnification factor of 25. The outer diameter of the fiber was 1.54 mm and the inner diameter was 0.64 mm. The slightly higher outer diameter (with respected to that reported for Figure 3 C in the previous paragraph) was either due to optical aberration or due to fiber expansion due to hydration. Fibers obtained by spinning and extrusion techniques generally have bends, twists, turns, and other types of imperfections. This is usually due to the flow processes involved in these techniques. Figures 3 and 4 clearly show that alginate fibers obtained using the molding technique had excellent definition and uniformity of appearance, and were straight. The inner and outer surfaces were smooth and the fibre shape was perfectly circular. The inner and outer diameter, and therefore the thickness of the fiber were uniform along the length of the fiber. A particularly attractive feature of this molding technique is that the alginate 125 fibers can be fabricated under sterile conditions without too much complication. Moreover, the fabrication was carried out under low pressure and stress conditions without the need for electric current. Also, the data shown in the previous paragraph clearly show the high degree to reproducibility with which the fibers could be molded. During alginate fiber fabrication by extrusion, the nascent fiber remains exposed to a large body of liquid and this increases the possibility of leakage of material being immobilized, i.e. cell or drugs. By contrast, during fiber fabrication using the hollow fiber membrane based molding technique no such exposure to bulk liquid takes place and consequently the loss of material are expected to be significantly lower. All these attributes would be particularly appealing in biomedical applications such as fabrication of inserts, soft delivery and connecting tubes, tissue engineering scaffolds and drugdelivery devices. The main disadvantage of the molding technique in its current form is that it is not suitable to producing large quantities of fibers. Also, the maximum length of the fiber is restricted to the length of the mold, i.e. the hollow fiber membrane. 126 Figure 5 Confocal laser scanning (CLS) microscopy images of the FITC-dextran alginate composite hollow fiber, (A) z-stack images obtained along the Z-axis at sequential depths of 200μm, (B) five times magnification, and (C) twenty-time magnification For imaging the interior of the fiber, FITC-dextran was added to the alginate solution prior to fiber fabrication. The sodium alginate concentration used in this experiment was 2.5 %, the FITC-dextran concentration was 2 mg/mL, the calcium chloride concentration was 1 M, an Accurel PP S6/2 membrane was used as mold, and the cross-linking was carried out for 60 seconds. The alginate hollow fiber thus produced was physically stiffer and more opaque than the fibers produced using sodium alginate alone. Most likely, dextran reinforced the structure of the alginate fiber and made it stronger. Figure 5 shows the images of the composite alginate fiber obtained by confocal laser scanning 127 microscopy. Figure 5A shows the z-stack images obtained along the axis at sequential depths of 200μm while B and C show respectively five-times and twenty-time magnified images of the same. The addition of the fluorescent additive facilitated a better view of the interior of the alginate fiber. The fiber had uniform circular cross-sectional area, with consistent in inner and outer diameter along the axis as evident from the z-stack images. The inner and outer surfaces of the fiber were smooth and the fluorescent additive was more or less uniformly distributed, except for a circular band of low fluorescent intensity close to the outer surface, and some intense fluorescent patches which were clearly visible under high magnification. Figure 6 Scanning electron microscopy (SEM) images of calcium alginate hollow fiber (400 X magnifications) fabricated with 2.5 % sodium alginate and 1 M calcium chloride, using Accurel PP S6/2 membrane as mold, with a cross-linking time of 60 seconds. 128 Figure 7 TEM image obtained with the hollow fiber described in Figure 6 at (A) 5000 X magnification, and (B) 75000X magnification Figure 6 shows scanning electron microscopy (SEM) images of calcium alginate hollow fibers (at 400 X magnifications) fabricated using 2.5 % sodium alginate, 1 M calcium chloride solution, Accurel PP S6/2 membrane as mold, and a cross-linking time of 60 seconds. The outer diameter of the hollow fiber under imaging condition was 560 μm while the wall thickness was 140 μm. Consistent with the earlier imaging techniques, the inner curved surface of the fiber was smooth and of perfectly circular curvature. The distortion on the outer surface was caused by the tweezers used to handle and process the fiber for imaging. The bulk material comprising the fiber appears to be homogeneous, but a skin-like structure is observed on the inner surface. Figure 7 shows the TEM image obtained with the same hollow fiber at two different magnifications (i.e. 5000X and 129 75000X respectively for A and B). The sponge-like porous structure of calcium alginate is evident at both levels of magnification. Figure 8 Effect of sodium alginate concentration on dimension of alginate fiber (mold: Accurel PP S6/2, calcium chloride concentration: 1 M; cross-linking time: 60 seconds) Figure 8 shows the effect of sodium alginate concentration on alginate hollow fiber dimension. These experiments were carried out using Accurel PP S6/2 membrane as mold with 1 M calcium chloride solution as cross-linker, the molding time being 60 seconds. While the dimension of the alginate hollow fiber did not show much responsiveness to sodium alginate concentration, there were significant differences in the strength and appearance of material obtained. The fiber obtained with 1 % alginate (data not shown in Figure 8) was quite fragile and difficult to measure due to its highly flexible 130 nature. The fiber obtained with 2.5% alginate was much more defined but the external surface was much rougher that that obtained using 5% sodium alginate solution. The strongest and most well defined fiber was obtained with 5% alginate. These fibers was also more opaque and of higher stiffness. Their slightly lower outer diameter was possibly due to dehydration during the gel formation, typically observed during crosslinking of highly concentrated polymer solutions [29]. Figure 9 Effect of calcium chloride concentration on dimension of alginate fiber (mold: Accurel PP S6/2, sodium alginate concentration: 5%, cross-linking time: 60 seconds) Figure 9 shows the effect of calcium chloride concentration on the dimension of the alginate fibers produced with 5% sodium alginate and 60 seconds of cross-linking time, using Accurel PP S6/2 membrane as mold. The cross-linker concentration had a far more 131 significant effect on the fiber dimension than the alginate concentration. The outer diameter increased very significantly when the calcium chloride concentration was decreased from 1 M to 0.5 M, and increased even further when the concentration was decreased to 0.1 M. The inner diameter also increased when the cross-linker concentration was decreased. However, the thickness increased with increase in crosslinker concentration. While the increase in hollow fiber wall thickness with calcium chloride concentration was due to greater cross-linker penetration into sodium alginate driven by greater concentration gradient, the effects on the inner and outer diameter are harder to explain. A plausible explanation is that the higher calcium chloride concentration caused shrinkage of the alginate structure by dehydration, leading to fibers of lower outer and inner diameters. 132 Figure 10 Effect of cross-linking time on dimensions of alginate fiber (mold: Accurel PP S6/2, sodium alginate concentration: 5%, calcium chloride concentration: 1 M) Figure 10 shows the effect of cross-linking time on the dimension of alginate fibers obtained using Accurel PP S6/2 membrane as mold with 5 % sodium alginate, and 1 M calcium chloride as cross-linker. While the outer diameter of the alginate fiber did not change appreciably; the inner diameter did decrease with increase in cross-linking time due to greater penetration of calcium ion within sodium alginate held within the mold. While the difference in inner diameter between 30 and 60 second cross-linking was not very significant, solid fiber was obtained by 600 seconds of cross-linking. 133 Figure11 Light microscopy image of HUVEC embedded alginate hollow fiber (image obtained one day after fabrication, magnification: 100 X, sodium alginate concentration: 1%, mold: Tetronic, calcium chloride concentration: 0.1 M, cross-linking time: 30 seconds) The feasibility of immobilizing cells was demonstrated by fabricating HUVEC embedded calcium alginate hollow fibers under sterile condition. The fabrication process was similar to that described in the previous paragraphs. Sodium alginate solution (2%) prepared in culture medium was mixed in 1:1 ratio with cells suspension. The mixture thus obtained was found to be less viscous than 1% sodium alginate solution prepared in water. This was possibly due to dilution caused by water released from cells due to osmotic effect. Tetronic hollow fiber membrane (0.8 mm i.d., 1.2 mm o.d.) was used as mold and cross-linking was carried out for 30 seconds using 0.1 M calcium chloride. Figure 11 shows the HUVEC embedded alginate hollow fiber obtained, the image being taken one day after fabrication by light microscopy using 100 X magnification. The cells 134 were uniformly dispersed and appeared to be viable. The cells appear bright in the center due to reflected light. Image analysis indicated that the density of immobilized cells was 4 × 106 cells /mL. The tissue or cell mass was not succeed during one week incubation period, HUVECs was not sufficient choice for immobilized system. Relatively HUVECs are sensitive on culture environment compared to fibroblast or neural cell line. Also the matrix composition can be stiff for the cell migration. For the future reference, the feasibility test can be conducted with microorganism or mammalian cells. The above results clearly demonstrate the suitability of the hollow fiber membrane based molding method for fabricating very well defined hollow or solid alginate fibers. The method as discussed is not suitable for large-scale production, but may be developed further for continuous extrusion of molded fibers. The technique as discussed in this paper will be particularly attractive in applications where small-scale precision fiber fabrication under sterile and gentle conditions is required. 5.5 Conclusion Calcium alginate fibers, both solid and hollow, could be fabricated by using a hollow fiber membrane based molding technique. Cross-linking agent (calcium chloride) held within the pores of the membrane diffused into the sodium alginate held within the membrane, cross-linking the alginate in an inward direction. The outer diameter of these fibers depended primarily on the inner diameter of the mold while the inner diameter 135 could be controlled based on the cross-linking time. Prolonging the cross-linking time resulted in the formation of solid alginate fibers. Factors such as alginate concentration and calcium chloride concentration were found to affect the dimension and physical properties of the fabricated fibers. The inner and outer surfaces of the hollow fibers were very well defined and the fibres were perfectly circular. The inner and outer diameter, and therefore the thickness of the fiber were uniform along the length of the fiber which were free from imperfections such as bends, twists, and turns and could be fabricated with high degree of reproducibility. The fibers could be fabricated under sterile condition and the technique did not involve the use of high pressure, high shear stress or electric current. The molding technique is therefore particularly appealing for producing inserts and soft tubes for biomedical applications, tissue engineering scaffolds and drug-delivery devices. However, the technique in its current form it is not suitable to producing large quantities of fibers and the maximum length of the fiber is restricted to the length of the mold. 136 5.6 References [1] A.S. Hoffman, A. Afrassiabi, L.C. Dong, Thermally reversible hydrogels: II. Delivery and selctive removal of substances from aquesous solutions, J. Control. Release. 4 (1986) 213-222. [2] G. Skjåk-Braek, Alginates: biosynthesis and some structure-function relationships relevant to biomedical and biotechnological applications, Biochem. Soc. T. 20 (1992) 27–33. [3] B. V. Slaughter, S.S. Khurshid, O.Z. Fisher, A. Khademhosseini, N.A. Peppas, Hydrogels in regenerative medicine, Adv. Mater. 21 (2009) 3307–3329. [4] S.J. Bryant, C.R. Nuttelman, K.S. Anseth, Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH / 3T3 fibroblasts in vitro, J. Biomater. Sci. 11 (2000) 439–457. [5] K.Y. Lee, D.J. Mooney, Hydrogels for Tissue Engineering, Chem. Rev. 101 (2001) 1869–1880. [6] K.I. Draget, G. Skjåk-Braek, O. Smidsrød, Alginate based new materials, Int. J. Biol. Macromol. 21 (1997) 47–55. [7] O. Smidsrød, G. Skjåk-Braek, Alginate as immobilization matrix for cells, Trends Biotechnol. 8 (1990) 71–78. [8] G.T. Grant, E.R. Morris, D.A. Rees, P.J.C. Smith, D. Thom, Biological interactions between polysaccharides and divalent cations: The egg-box model, FBES Lett. 32 (1973) 195–198. [9] Y. Yeo, K. Park, Control of encapsulation efficiency and initial burst in polymeric microparticle systems, Arch. Pharm. Res. 27 (2004) 1–12. [10] X. Huang, C.S. Brazel, On the importance and mechanisms of burst release in matrix-controlled drug delivery systems, J. Control. Release. 73 (2001) 121–136. [11] R.H. Li, D.H. Altreuter, F.T. Gentile, Transport characterization of hydrogel matrices for cell encapsulation, Biotechnol. Bioeng. 50 (1996) 365–373. [12] R. Zarzycki, Z. Modrzejewska, K. Nawrotek, Drug release from hydrogel matrices, Eco.Chem. Eng. 17 (2010) 117–136. [13] N. Doshi, B. Prabhakarpandian, A. Rea-Ramsey, K. Pant, S. Sundaram, S. Mitragotri, Flow and adhesion of drug carriers in blood vessels depend on their 137 shape: a study using model synthetic microvascular networks, J. Control. Release. 146 (2010) 196–202. [14] Y. Dzenis, Material science. Spinning continuous fibers for nanotechnology, Science. 304 (2004) 1917–1919. [15] D.H. Reneker, I. Chun, Nanometre diameter fibres of polymer, produced by electrospinning, Nanotechnology. 7 (1996) 216–223. [16] M. Bognitzki, W. Czado, T. Frese, A.Schaper, M. Hellwig, M. Steinhart, A. Greiner, J.H. Wendorff, Nanostructured fibers via electrospinning, Adv. Mater. 13 (2001) 70–72. [17] C.A. Bonino, M.D. Krebs, C.D. Saquing, S.I. Jeong, K.L. Shearer, E. Alsberg, S.A. Khan, Electrospinning alginate-based nanofibers: From blends to crosslinked low molecular weight alginate-only systems, Carbohydr. Polym. 85 (2011) 111–119. [18] D. Fang, Y. Liu, S. Jiang, J. Nie, G. Ma, Effect of intermolecular interaction on electrospinning of sodium alginate, Carbohydr. Polym. 85 (2011) 276–279. [19] J.J. Chang, Y.H. Lee, M.H. Wu, M.C. Yang, C.T. Chien, Preparation of electrospun alginate fibers with chitosan sheath, Carbohydr. Polym. 87 (2012) 2357–2361. [20] L. S. Barbarash, J. L. Elgudin, V. V. Sevostyanova, A. S.Golovkin, Tissueengineered vascular graft and its fabrication approach (2013) WO 2013151463 A2 [21] N. Traitcheva, H. Berg, Electroporation and alternating current cause membrane permeation of photodynamic cytotoxins yielding necrosis and apoptosis of cancer cells, Bioelectrochemistry. 79 (2010) 257–260. [22] N. Bhattarai, Z. Li, D. Edmondson, M. Zhang, Alginate-based nanofibrous scaffolds: structural, mechanical, and biological properties, Adv. Mater. 18 (2006) 1463–1467. [23] T. Takei, S. Sakai, H. Ijima, K. Kawakami, Development of mammalian cellenclosing calcium-alginate hydrogel fibers in a co-flowing stream, Biotechnol. J. 1 (2006) 1014–1017. [24] T. Takei, S. Yamaguchi, S. Sakai, H. Ijima, K. Kawakami, Novel technique for fabricating double-layered tubular constructs consisting of two vascular cell types in collagen gels used as templates for three-dimensional tissues, J. Biosci. Bioeng. 104 (2007) 435–438. 138 [25] T. Takei, N. Kishihara, S. Sakai, K. Kawakami, Novel technique to control inner and outer diameter of calcium-alginate hydrogel hollow microfibers, and immobilization of mammalian cells, Biochem. Eng. J. 49 (2010) 143–147. [26] M. Hu, R. Deng, K.M. Schumacher, M. Kurisawa, H. Ye, K. Purnamawati, J.Y. Ying, Hydrodynamic spinning of hydrogel fibers, Biomaterials. 31 (2010) 863– 869. [27] H. Onoe, T. Okitsu, A. Itou, M. Kato-negishi, R. Gojo, D. Kiriya, K. Sato, S. Miura, S. Iwanaga, K. Kuribayashi-shigetomi, Y.T. Matsunaga, Y. Shimoyama, S. Takeuchi, Metre-long cell-laden microfibers exhibit tissue morphologies and functions, Nat. Mater. 12 (2013) 584–590. [28] S.J. Shin, J.Y. Park, J.Y. Lee, H. Park, Y.D. Park, K.B. Lee, C.M. Whang, S.H. Lee, “On the fly” continuous generation of alginate fibers using a microfluidic device, Langmuir. 23 (2007) 9104–9108. [29] T.R. Cuadros, O. Skurtys, J.M. Aguilera, Mechanical properties of calcium alginate fibers produced with a microfluidic device, Carbohydr. Polym. 89 (2012) 1198–1206. [30] J. Su, Y. Zheng, H. Wu, Generation of alginate microfibers with a roller-assisted microfluidic system., Lab Chip. 9 (2009) 996–1001. [31] S. Sugiura, T. Oda, Y. Aoyagi, M. Satake, N. Ohkohchi, M. Nakajima, Tubular gel fabrication and cell encapsulation in laminar flow stream formed by microfabricated nozzle array, Lab Chip. 8 (2008) 1255–1257. [32] J. Yan, Y. Huang, D.B. Chrisey, Laser-assisted printing of alginate long tubes and annular constructs., Biofabrication. 5 (2013) 1-8. [33] R.T. Weitz, L. Harnau, S. Rauschenbach, M. Burghard, K. Kern, Polymer nanofibers via nozzle-free centrifugal spinning, Nano Lett. 8 (2008) 1187–1191. [34] T. Boland, T. Xu, B. Damon, X. Cui, Application of inkjet printing to tissue engineering., Biotechnol. J. 1 (2006) 910–917. 139 Chapter 6 Contributions and Recommendations The following was achieved through the research work described in this thesis. 6.1 Monoclonal Antibody Polishing Process Deign Based Membrane Chromatography PVDF membranes were studied for environmentally responsive characteristic under salt concentrations [1- 5]. This membrane was suggested as bioseparation tool using different apparent hydrophobic interactions between protein and membrane matrix. IgG and its aggregates [1], and rabbit IgG and protein-A conjugated rabbit IgG [2] were used as model proteins for assessing subtle hydrophobic difference. Based on earlier studies, PVDF membrane chromatography was utilized for designing the monoclonal antibody polishing process. The model proteins were choose according to industry practice, which were monoclonal antibody, di-, tri-mer antibody aggregates, and conjugates with proteinA. Individual protein hydrophobic interaction with PVDF membrane chromatography was investigated, and then feed solution (mixture of three proteins) was applied on hydrophobic interaction membrane chromatography with optimized operating conditions. A polishing step was built up with flow-through mode that allowed product (monoclonal antibody) to flow through the membrane chromatographic system and two impurities (aggregates and protein-A conjugates) were trapped in it. 140 This case study could afford many opportunities in the last step of impurities removal of monoclonal antibody production line as a polishing step. It is suitable for fewer amounts of impurities bound on membrane with high flow rate. It is even more beneficial from an industries perspective, as the process does not need to be stopped in the middle to switch buffers, because the fluid with final product will be collected in flow through stream. The membrane chromatography adsorbent can be used up by calculating the amount of impurities reached up to 80% of membrane binding capacity. This membrane adsorbent can be regenerated by lowering salt concentration if it is necessary. The second contribution would be the feasibility of this process development applied in other biopharmaceutical products. As chapter 1 listed the advantage of membrane chromatography, the biopharmaceutical product process development will be guided toward to membrane chromatographic separation. With relative less membrane chromatography technologies established time compared to resin-based column chromatography, the growth of membrane chromatography performance is fast enough which will be applied in biopharmaceutical production line. For the future work can be suggested below. Membrane binding capacity can be improved by adding hydrogel coating/grafting on the membrane substrate. 141 6.2 inexpensive membrane chromatography with paper based composite membrane The development of a PVCL composite membrane which is applied to membrane chromatography has been assessed. In earlier work [6], the interests of composite membrane were more focused on protein adsorbent. In this thesis chapter 3 achieved PVCL composite membranes were adopted as a protein separation tool using its environmentally responsive behaviour. Two major plasma proteins (hIgG and HSA) and the popular hormone (insulin) were choosing for model protein. It flowed into the composite membrane chromatography system at 1.3M of ammonium sulphate concentration. First, the least hydrophobic protein, HSA, passed through the membrane matrix without interaction. The remaining two proteins, hIgG and insulin, were eluted by reducing salt concentration incrementally. From this work, “custom made” hydrogel coated on filter paper membrane with an inexpensive fabrication price, controllable of cross-linking degree, and flexible geometries could be benefit both manufacturing and research areas. For comparison for the economical aspect in chapter 2 and 3, commercially available hydrogel grafted membrane (PVDF) for membrane chromatographic application increases the total cost of production. For the future work can be suggested below. PVCL hydrogel stability needs to be improved over period of time. Composite membrane has to be stored in liquid solution for the prevention of dehydration which led breakage of polymer chain. 142 Protein fouling problem was presented after repeating separation runs. Several experimental runs indicated pressure increases due to the protein fouling. 6.3 Hollow fiber membrane modification with natural hydrogel The fouling proteins or biomolecules on hollow fiber membrane surface is the chronic problem in membrane bioreactor and water treatment applications. Due to the concentration polarization, the mass transport gradually lowers in comparison to initial operating conditions. The initial motivation behind his project was prevent biological (cell or cell debris) substance fouling/blocking on extra-capillary space, as it hindered mass transport of nutrients and metabolite counter diffusion in membrane bioreactor. A calcium alginate hydrogel was coated on single hollow fiber membrane for investigating hydrophilized membrane surface which is friendlier to cell adhesion but lower fouling behaviour. There are three points of novelty in this work. The first is that the fabrication technique of calcium alginate hydrogel is new in that cross-linking agents were stored within membrane pores and diffused out to sodium alginate in controlled manner. This process leads to the calcium alginate hydrogel coating being uniform and smooth along the entire membrane length. Second, ethanol facilitates calcium chloride solution, showing different hydrogel morphologies while having a sterile effect for biomedical applications. Third, changes of hydrophilic characteristic membrane surface had positive effect on water permeation, and DMSO mass transfer. This work has been investigated on each of these 143 individual components. It has much potential to be used in biomedical or biopharmaceutical industry. For the future work can be suggested below. Biocompatibility test can be obtained with cells (i.e. T-cells, bacterial for certain drug productions). In this project, only ultrafiltration hollow fiber membrane was test. Microfiltration hollow fiber membrane also can be a good candidate. Hydrophobicity can be varying by different membrane materials such as polyethersulfone, polysulfone, polypropylene, and PVDF hollow fiber membrane. 6.4 Calcium alginate fiber fabrication using hollow fiber membrane Alginate hollow fibers are commonly produced by a complicated experimental set-up and a large amount of samples (including immobilized cells). Since the size of set-up the fabrication process is large, it is difficult to keep in sterile condition. Some of these techniques, particularly electrospinning and two nozzle extrusion, also expose the cells to harsh conditions, leading to an increased cell death rate. In this work we present a novel method for fabricating calcium alginate on hollow fiber membranes. The membrane was not used as typical separation barrier; it was adopted as a mold where it provides the reservoir for cross-linking agents. Using this manner, it is easy to control the dimension such as diameter, length, thickness of fibers, and morphology of fiber can be manipulated by concentration of sodium alginate, cross-linking agent, and cross linking duration. 144 This opens up a new scope in membrane usage in tissue engineering area. In this thesis, only the fabrication process was obtained with different sizes of fibers and several characterizations of these fibers were performed with various imaging techniques. Biocompatibility test was obtained with human umbilical vein endothelial cells. For the future work can be suggested below. Further research involving cell immobilization (i.e. using human urinary bladder cell lines, epithelial cells, or human microvascular endothelial cells) Biodegradation rate and cell growth rate within calcium alginate fiber matrix must be undertaken for future work. Seeding method can be embedded (chapter 5) or spreading on the surface of calcium alginate fiber. Cell adhesion molecule can be treated on the surface such as gelatin, collagen or serum. In vitro experiments would check for functionality, leading to potential in vivo (mouse) experiments. 145 6.5 References [1] L. Wang, J. Hale, R. Ghosh, Non-size-based membrane chromatographic separation and analysis of monoclonal antibody aggregates, Anal. Chem. 78 (2006) 6863-6867 [2] X. Sun, D. Yu, R. Ghosh, Study of hydrophobic interaction based binding of immunoglobulin G on synthetic membranes, J. Membr. Sci. 344 (2009)165-171 [3] L. Wang, R. Ghosh, Purification of humanized monoclonal antibody by hydrophobic interaction membrane chromatography, J. Chromatogr. A 1107 (2006) 104-109. [4] R.Ghosh, Separation of human albumin and IgG by a membrane-based integrated bioseparation technique involving simultaneous precipitation, microfiltration and membrane adsorption, J. Membr. Sci. 237 (2004) 109-117 [5] X. Sun, D. Yu, R. Ghosh, Study of hydrophobic interaction based binding of immunoglobulin G on synthetic membranes, J. Membr. Sci. 344 (2009) 165-171. [6] K.Z. Mah, R. Ghosh, Paper-based composite lyotropic salt-responsive membranes for chromatographic separation of proteins, J. Memb. Sci. 360 (2010) 149–154. 146