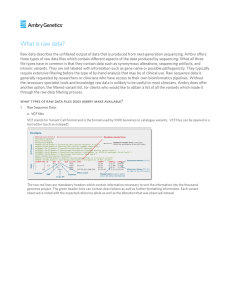
Supplementary data: Methods: Sequencing libraries were generated
advertisement

Supplementary data: Methods: Sequencing libraries were generated from genomic DNA using the Nextera Rapid library construction kit and enriched using the TruSight One capture panel (Illumina, San Diego, CA, USA). Libraries were sequenced on an Illumina MiSeq using version 3 SBS chemistry with a paired-end 150bp read length with dual indexing. Sequence data were streamed to Illumina BaseSpace and analysed using Illumina’s Enrichment application to generate BAM and vcf files. Annotation was added to the vcf data using Variant Analysis Studio, and exported into Excel for further analysis. The fastq files generated by the MiSeq were aligned to the human reference GRCh37/hg19 Variants were identified using the UnifiedGenotyper from GATK. The identified variants were recalibrated using GATK, and functional annotation of the vcf files was performed using VEP (ensembl 72)1, dbSNP137, the 1000 Genomes project2 for population allele frequencies, and the NHLBI GO Exome Sequencing Project3, using vcf-tools4. Results: Only one variant was found in the REN gene which was non-pathogenic (rs2272237, intronic, Global MAF C=0.2245/489). Looking at the genetic locus described by Piret et al.[11] (cytogenetic location 2p22.1-p21, genomic coordinates [GRCh37]: 2:38,600,0047,800,000), 721 variants were found in this region, of which 166 variants had a frequency of <1%. From the rare variants (<1%) only 9 were exonic and they are splicing variants with no amino acid changes. After cross-checking with dbSNP and EVS (exome variant server) databases, four of the splicing variants were found to be too frequent. The remaining five variants were not found to be candidates for a pathological mutation. References for supplementary data: 1. McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. BMC Bioinformatics 2010, 26(16):2069-70 2. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA and 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012 Nov 1; 491(7422):5665. doi: 10.1038/nature11632. 3. http://evs.gs.washington.edu/EVS/ Last accessed 31st March 2014. 4. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker R, Lunter G, Marth G, Sherry ST, McVean G, Durbin R and 1000 Genomes Project Analysis Group. The Variant Call Format and VCFtools. Bioinformatics, 2011.