Supplementary Methods The microarray and hybridization The Lund
advertisement

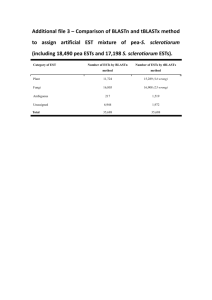
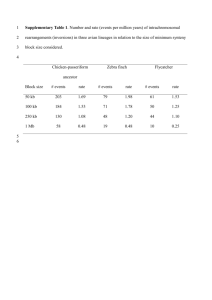
Supplementary Methods The microarray and hybridization The Lund-zfa array is a custom Affymetrix array produced for the zebra finch [1]. It contains approximately 22600 ESTs, representing approximately 15800 non-redundant genes, and each EST is represented by 11 (25 bases long) probes (except for 148 ESTs that are represented by 8-10 probes). In total, there are 254430 probes on the array. The array contains no Affymetrix mismatch probes. High-quality total-RNA samples representing each individual (the 40 zebra finch samples) were delivered to the Swegene Center for Integrative Biology at Lund University (SCIBLU genomics, http://www.lth.se/sciblu), Lund, Sweden, where they were hybridized according to standard Affymetrix protocols for RNA (see Naurin et al. [1,2] for a detailed description of the hybridization procedures). Signal intensities were normalized and filtered as previously described and to ensure high quality signal intensity measurements, we excluded ESTs that had not retained all 11 probes in a previous comparative genome hybridization analysis [cf. 1,2]. EST annotation and chromosome position Estimations of redundancies amongst the ESTs on the array had previously been made using the annotations produced for each sequence in the ESTIMA: songbird build 2 assembly [1,3]. Here, we used BLAST analyses against the 3.2.4-build of the zebra finch genome (http://www.ncbi.nlm.nih.gov/genome/guide/finch/) to generate annotation of chromosome and position, which in turn were used to remove redundancies and to compare autosomal and Z-chromosome gene expression. We used a cut off E-value of 10-10 for significant hits, and included only single hits (when more than one significant match occurred the best hit had to be <10-10 and the next nearest hit had to be more than 10-10 weaker) and hits separated by at least 5 kbp in the zebra finch genome. Our analyses are based on 10695 ESTs (10131 autosomal; 564 Z-linked). Data are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.ps515. Linear mixed models Gene expression (log2 signal intensity) was analysed with linear mixed models in the R package nlme [4]. The model included three fixed factors (‘sex’: male or female; ‘chromosomal location’: autosomal or Z-linked; ‘inbreeding treatment’: inbred or outbred) and their interactions. To account for array effects, ‘individual identity’ was included as random factor. Significances of the fixed factors and their interactions were calculated with F-tests using the anova option in R. The denominator degrees of freedom as shown in Table 1 are upper bounds. There is no ‘correct’ denominator degrees of freedom because the F-test is an approximation of the mixed model, but since the real degrees of freedom should be lower than the upper bound the present test is conservative [4, 5]. Note also that the fixed effect of main interest in the present study, i.e. inbreeding treatment, was consistently associated with very low F-values (Table 1). 1. Naurin S, Hansson B, Hasselquist D, Kim YH, Bensch S. 2011 The sex-biased brain: sexual dimorphism in gene-expression in two species of songbirds. BMC Genomics 12, 37. 2. Naurin S, Bensch S, Hansson B, Johansson T, Clayton DF, Albrekt AS, von Schantz T, Hasselquist D. 2008 A microarray for large-scale genomic and transcriptional analyses of the zebra finch (Taeniopygia guttata) and other passerines. Mol Ecol Res 8, 275-281. 3. Replogle K, Arnold AP, Ball GF, Band M, Bensch S, Brenowitz EA, Dong S, Drnevich J, Ferris M, George JM, et al. 2008 The Songbird Neurogenomics (SoNG) Initiative: community-based tools and strategies for study of brain gene function and evolution. BMC Genomics 9, 131. 4. Pinheiro J, Bates D, DebRoy S, Sarkar D and R Core Team. 2014 nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1-117, http://CRAN.Rproject.org/package=nlme. 5. Bates DM. 2005 Fitting linear mixed models in R. R News 5, 27–30.