Lecture 30: Molecular interactions
advertisement

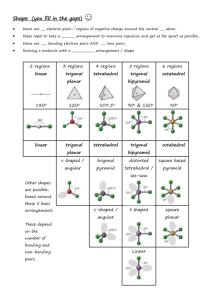
Lecture 30: Molecular interactions Review o Molecular orbitals o Hybridization Today o Bonding in inorganic transition metals o Geometric interactions o Bond stretching/bending o Intramolecular rotations o Charge dependent nonspecific interactions o Coulombic interactions o Dipolar interactions o London-Van-der Waal interactions Bonding in inorganic complexes Here we are mainly concerned with the geometric disposition of ligands surrounding transition metal ion focusing on how the d-orbitals interact with ligands. Clearly, energies of degenerate orbitals will be enhanced or depressed depending on the geometry of the complexes. In addition, the extent of energy level splitting will depend on the charge on metal ions and ligands. A series of ligands are shown below rated according to the strength of binding. The strength of ligand binding interaction has important consequences in paramagnetic properties of the transition metal ions. What is the spin state iron in octahedral complexes? Iron atom in octahedral geometry has its five d-orbitals split as shown below. The structure on the left has a lower splitting so the fiveelectron configuration is such that all the electrons occupy separate orbitals. Of course, this involves an energy cost; 2E, but this compensated for by the reduction in the electron-electron repulsion interactions. This is because H2O is a weak ligand. On the other hand, if we consider a strong ligand such as CN-, the splitting on the two sets of energy levels is greater. Now the minimum energy configuration of the system involves electron pairing as shown in the right figure. These simple energetic considerations have dramatic influence on the magnetic properties of the complex. The high spin complex, in the left figure, has all the spins unpaired hence the Fe in this state exhibits strong paramagnetism, which is directly related to the number of unpaired electron present in a molecule. Intra-molecular geometric interactions As we discussed in the context harmonic oscillator problem, stretching or compression of bonds can be energetic costly. If we consider a simple Hooks law for stretching a single or a double bond we find that for low quantum numbers. Similarly Hooks law is a good approximation for the bond bending in a three-carbon atom system as shown below. However energies involved are much smaller. Owing to zero point energy the contributions from the vibrational modes cannot be simply ignored. Rotation around a bond Just as hooks law potential is a good approximation for the bond bending/stretching, simple sine wave potential is a good approximation for the rotation around single bond Although the energy minima around carbon bond are shown above to have identical energy at minimum, in reality, the trans configuration generally has lower energy. The structure of the potential also critically depends on pendent groups attached. Rotation around a double Bond Similarly, rotation around a double bond can be considered. Since the rotation requires breaking a pi bond the energies needed to overcome the potential barriers are higher, amides for example show: This rotation is of importance for the geometric stability of protein superstructure. Such empirical formulae for the potential are extremely useful in the calculation of minimum energy structures, for example in the molecular mechanics calculations. Non-covalent interactions: Coulomb interaction Coulomb potential between two charges is given by: As we have seen before, the molecular orbital method allows us to calculate the probability density at a given atom depending on which molecular orbitals are occupied. As an example consider the following molecule: Thus to calculate coulomb energy between to such molecules we have to carry out a sum over all the binary interactions between charges on both molecules. Peter Debye who suggested we should also consider the dipole moment of such a molecule introduced great simplification. It is defined as q.r where q is the average positive charge located at distance r from the average –ve charge. This dipole moment accounts for the non-centro-symmetric distribution of charges in molecules. Dipoles To calculate the net dipole moment of molecule we simply evaluate sum of q.x,q.y,q.z for all atoms in molecule. For the above molecule: Since the dipole moment is a vector we can calculate the magnitude and the orientation of the dipole moment from the standard vector algebra: 2x y2 tan y y Fundamentally, dipole moment characterizes the polarity of molecules. For centrosymmetric molecules the net dipole moment vanishes, for example CH4 or CCl4. One advantage of this concept is that the dipole moment can be experimentally measured using a capacitance measurement technique. The difficulty in applying such concept to real biological molecules is the effect of solvent. Dipole-dipole interactions. The interaction between the two dipoles is given by: Here, R12 is the distance between the two dipoles. Since the dipoles are vectors we have to consider their dot products. This is because the potential is a measure of energy, which is a scalar quantity. The dipolar interaction decreases as the distance between the dipoles is increased (-3 rd power law). The contribution of the dipolar energy can be positive, negative or zero, depending on the relative orientation of the dipoles. Consider following examples: London interaction: Induced dipole-dipole interaction This is important where polarizable molecules (such as benzene) are involved. Presence of a strong dipole in the vicinity of a polarizable molecule leads to an induction of dipole moment. This interaction is always negative and was studied by London: Notice the interaction is much shorter range than the dipoledipole interaction we considered previously; since it varies inversely as the sixth power with respect relative separation of molecules. Furthermore as r0 this interaction decreases dramatically, suggesting a molecular fusion. It was van der Waal who advocated an even a shorter-range repulsive interaction with 1/r12 dependence to counter the effect of London interaction. Thus, the net interaction, London+Van der Waal, looks like as shown in the figure to the right. This is the famous 6-12 potential. The position of the minimum gives the Van der Waal radius of a molecule.