Lecture 3, Solid-Water Interface
advertisement

1
GES 166/266, Soil Chemistry
Winter 2000
Lecture Supplement 3
Solid-Water Interface
VI.1 SURFACE CHARGE
VI.1-1, Permanent Charge
There are two types of charge generally associated with mineral and organic
surfaces: permanent and variable charge. As we have already stated, permanent charge
arises from isomorphic substitution within a mineral. The substitution results in a charge
deficiency which is delocalized so that we can think of this charged as distributed across a
surface plane. In the phyllosilicate minerals it is the (001) plane that dominantly exhibits
the permanent charge. An important point to consider is that although a charge was
created from the ion substitution no bonds were altered; this means that the (001) plane,
while having an excess charge, does NOT have a chemical affinity for solution ions. It
only wishes to satisfy its electrical charge. As a consequence, ions will be bound only
through electrostatic forces to the permanently charged (001) surfaces of the
phyllosilicates. A few important points to remember about permanent charge:
1. it is pH independent
2. it is developed by isomorphic substitution
3. it is represented by the charge symbol o
VI.1-2, Variable Charge
Unsatisfied bonds at the terminal ends of minerals and organic matter result in a
surface charge. These surfaces, however, have very different properties from those
discussed for the permanently charged surfaces. Firstly, as the name implies, the charge
can vary depending on the solution conditions; this charge is primarily a function of pH
and is sometimes referred to as pH dependent charge. This results from the surface
oxygens of the hydrous oxides and many organic functional groups, such as the
carboxylic acid groups, having a high affinity for H+ ions. The addition or release of
protons from the surface results in different charges. For example, consider the
protonation reactions of an iron oxide surface functional group (surface site).
>Al-OH-1/2 + H+ <==> >Al-OH2+1/2
>Al2=O-1 + H+ <==> >Al2=OHo + H+ <==> >Al2=OH2+
In this reaction, the species to the left would represent a very high pH condition, the
middle a moderate pH, and the right species a low pH. As you can see, the surface charge
changes with pH not only in magnitude but also in sign. Note that at low pH values the
2
surface is positively charged, as the pH increases the positive charge is reversed to a
negative charge which increases with further increased pH.
Because the charge can change signs, we need to introduce another important
term: The zero point of charge (ZPC). The ZPC is the solution pH at which the NET
surface charge is zero. This does not mean that the surface has no charge, but rather that
there are equal amounts of positive and negative charge. At pH values below the ZPC,
the surface has a positive charge while it has a negative charge at pH values above the
ZPC.
A second difference between permanently and variably charged surfaces is that on
the latter the charge is localized at specific sites. In fact, adjacent sites may even have
opposing charges. A third, and very important, difference is that variable charge surface
sites are chemically reactive. In response, ions may be retained on the surface through
electrostatic attractive forces or through a chemical bond. Summarizing important
characteristics of variable charged surfaces:
1. they are pH dependent
2. developed from terminal bonds
3. represented by H
4. surface groups do NOT develop a charge greater than |1|
5. dominant surfaces: hydrous metal-oxides, kaolinite, and organic matter
6. surface groups are weak acids and be defined by their ionization parameter()
Other Planes of Charge that can occur in the solid/water interface are attributed to: Innersphere ions (is), outer-sphere ions (os), and the diffuse Layer (d). o and H are the
'mineral' charge while the other 3 planes are due to reactions from the solution. It is
important to note that the diffuse layer counter balances all the other planes of charge.
The zero point of charge (ZPC) is defined as the pH where: o + H + is + os = 0.
This definition also means the ZPC is where the total particle charge is zero: (+) = (-).
Some important points about the ZPC to remember are: (i) flocculation is greatest at pH =
ZPC, (ii), mineral dissolution is at a minimum when pH = ZPC, and (iii) soils tend to
weather toward a pH = ZPC.
VI.2 ION RETENTION
Unlike organic molecules, inorganic species cannot be degraded. They can,
however, be retained on mineral surfaces or form discrete precipitates; in either case they
are removed from the mobile aqueous phase and their bioavailability is consequently
restricted. Retention processes thereby decrease the risk imposed by contaminants but
they also can decrease the availability of needed plant nutrients. Accordingly, it is
important for us to understand the processes by which ions are removed from solution
and to have an idea of how strongly the ions will be immobilized. Or, in other words,
what is the potential for the ions to be released back into solution?
3
Ions can bind by different mechanisms (reactions); the retention strength is
dependent on mechanism. Additionally, models predicting ion sorption will differ
depending on the mechanism. Possible retention mechanisms include adsorption and
surface precipitation.
Before proceeding further we should define some terms so that we will all be
speaking the same language. The terms sorption, adsorption, absorption, precipitation,
surface precipitation, retention, and others are all used to refer to the loss of a species
from solution. They differ, though, in their implicit meaning. The definitions we will
use are as follows.
VI.2-1, Sorption Terms
Sorption: The retention of a species without implication to its retention mechanism. This
term is inclusive of adsorption, absorption, precipitation, and surface precipitation.
Adsorption: The binding of an ion or small molecule to a surface at an isolated site--a 2D surface complex. There is no interaction (or at least only minimal interaction) between
adsorbed species.
Absorption: The uptake of a species WITHIN another material. This mechanisms is
somewhat analogous to water uptake into a sponge.
Surface precipitation: A 3-dimensional growth mechanism of a species on a surface.
This mechanism differs from adsorption in that the retained species directly interact with
each other on the surface and can even have the solid structure grow away from the
original substrate.
Precipitation: The formation of a 3-D structure without the association of a substrate
(sorbent) material. This process occurs in solution directly and leads to discrete particles
(it is also refereed to as a 'homogeneous precipitate').
Sorptive: A species in solution that may undergo sorption.
Sorbate: A species retain on another material.
Sorbent: The substrate material responsible for the retention of a solution species.
As these definitions should imply, there are many different processes responsible for the
removal of a species from solution. We now need to look at these various mechanisms in
more detail to gain an understanding of their retention strengths.
4
VI.2-2, Adsorption Mechanisms
The energy of adsorption can be divided into two components, that from the
electrostatic interaction and that from the chemical: Adsorption Energy = Eelectrostatic +
Echemical. It is important to note that even if the electrostatic component is negative, the
chemical affinity of an ion for the surface can override it. That means ions can adsorb
against an electrostatic gradient, e.g., transition metal cations bind to goethite at pH < 8.
We can separate possible adsorption mechanisms into three classes:
1) Inner-sphere complexation (chemical reaction)
- a chemical reaction between the surface and the ion
- 'specific adsorption'
- very strong association
- exchangeable
2) Outer-sphere complexation (electrostatic reaction)
- localized electrostatic charge neutralization
- 'non-specific' adsorption
- exchangeable
- analogous to ion pairs
3) Retention in the Diffuse Swarm (electrostatic attraction)
- delocalized electrostatic attraction
- 'swarm' neutralizes remaining surface charge
- exchangeable
VI.2-3, Chemical Surface Reactions:
When we discussed the charge developed from broken bonds we recognized an
imbalance in charge that resulted at a surface. In addition to the charge imbalance, these
surface functional groups are also coordinately unsaturated and would therefore like to
satisfy their bonding environments. We should realize then that these groups or surface
sites may incorporate other ions from solution into their structure. When this happens the
adsorbed ion will loose its hydration sheath and a chemical bond (covalent or ionic) will
result. As might be expected, these bonds are much stronger than those of an outersphere complex and we generally do not consider such sorbates as exchangeable. Innersphere complexes do not form on the (001) plane of the phyllosilicates but are readily
formed on the (010) and (100) plane of the phyllosilicates; in fact, all of the variable
charged surfaces allow inner-sphere complex formation.
In addition to considering the surface we should also discuss the ions which may
form inner-sphere surface complexes. It is beyond the scope of this course to present a
rigorous discussion about electron characteristics of an ion which dictates the type of
complex it forms, but there are some general rules we can assign to qualitatively assess
this phenomena. Generally, the alkaline earth ions will form outer-sphere complexes
only, while the transition metals have the capacity to form inner-sphere complexes. We
can not generalize about the ions from the right side of the periodic chart as this is
5
partially dependent on their molecular arrangement; here is a list of their capacity to form
inner-sphere complexes (realize, of course, that any charged ion has the capability to be
retained due to electrostatic forces as well).
Ion
ClSO42NO3FPO43SeO32SeO42AsO43AsO33CrO42-
Inner-sphere
Capacity
no
partially
no
yes
yes
yes
partially
yes
partially
yes
VI.2-4, Exchange Reactions
A significant means by which ions adsorb is due to an electrostatic attraction
between an ion and a surface of opposite charge. Electrostatic reactions occur between
any ion and surface of opposite charge. Such electrostatic forces may arise from the
permanent negative charge of a phyllosilicate clay mineral and a cation such as Na+,
Ca2+, or many others. Remember that a variable charged surface may have either a
positive or negative charge (or both) depending on the solution conditions. Electrostatic
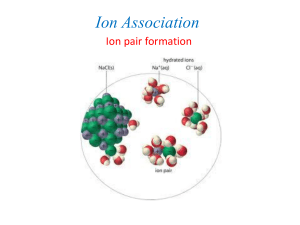
interactions between a surface and an ion are analogous to ion pairs that we discussed in
the Aqueous Chemistry portion of the course. As such, you should also remember that
this would be an outer-sphere complex where the surface and the ion maintain their
hydration sheath. These outer-sphere complexes are rather weak compared to chemical
forces and result in an exchangeable sorbate. In fact, it is predominantly the
electrostatically bound ions that make up the cation exchange or anion exchange capacity.
Electrostatically bound ions can be displaced by other ions or displaced simply due to a
diffusion gradient. Exchangeable ions are essential for maintaining plant nutrient levels,
but are not strong enough to immobilize environmental pollutants.
6.2-4.1, Cation Exchange Capacity
The CEC is usually dominated by Ca, Mg, Na, K, and Al; thus,
CEC (mmol charge) ≈ 2[Ca] + 2[Mg] + [K] + [Na] + 3[Al]
The selectivity of cation by exchanger is based on the ion's charge/size
6
1. SIZE: The smaller the hydrated radius the greater the affinity
(note: ions with small dehydrated radius have large hydrated)
2. VALENCE: This is the dominant factor influencing adsorption. The higher the
valence the greater the exchanger preference: 4+ > 3+ > 2+ > 1+.
The preference of an ion for a surface is summarized by the Lytropic Series (strength of
retention):
Th4+ > Al3+ > La3+ > Ba2+ ≈ Sr2+ > Ca2+ > Mg2+ ≈ Cs+ > Rb+ > NH4+ > K+ > Na+ ≈ Li+
However, deviations from Lytropic series occur if specific chemical affinity occurs.
Examples of such deviations include:
1. K+ on vermiculite
2. Increased affinity of highly charged surfaces for highly charged ions
3. Vermiculite also has an unusually high affinity for Mg
VI.2-5, Precipitation Mechanisms
Precipitation reactions result from a solution being oversaturated with respect to a
mineral phase. Solubility constants for precipitation in bulk solution are tabulated in
many text books. Using these constants, one can use the saturation index we discussed
previously to determine if a solution is undersaturated (SI < 0) , oversaturated (SI > 0), or
in equilibrium (SI = 0) with a solid,
SI = log (IAP / Ksp)
where IAP is the ion activity product for the specific reaction and Ksp is the solubility
constant for this reaction. Remember that IAP is the measured activity values for the
reaction while Ksp is the value representative of an equilibrium situation; they are only
equal if the system is at equilibrium (SI = 0).
While the SI gives a convenient means for assessing the thermodynamic
possibility of precipitation, it does not tell us whether the reaction will actually happen-only if it is possible. Kinetic factors usually govern the phase we can expect to form over
a short period of time. This is primarily dictated by the activation energy, or energy
barrier, of a reaction. Generally, large, well-crystallized particles have a lower Ksp but
higher activation energy. Consequently, we frequently find many amorphous particles in
soils due to their meta-stable conditions. Given sufficient time, these amorphous phases
will transform into more crystalline solids, which are thermodynamically more stable.
All this is applicable for the bulk solution, but what about the mineral/solution
interface? Here, there are additionally forces that must be considered. Unfortunately, it
is not yet possible to provide a quantitative value for most of these factors, so we must
restrict or discussion to a qualitative one. Due to electrostatic and chemical forces, Ksp
7
values are always lower in the interfacial area relative to the bulk solution. That is,
surfaces will catalyze the precipitation of solids. They will also partially influence the
mineral phase that forms. Consequently, although IAP values may indicate that the bulk
solution is undersaturated with respect to a mineral, such a mineral may form at the
solid/solution interface. Since this phenomena can not be quantitatively describe, it is
simply your job to remember that their is a potential for such a reaction.
Precipitation is modified relative to solution precipitation in 2 ways: (i) surface
lower activation energy and catalyzes precipitation (ii) electrostatic charge decreases Ksp
(thermodynamic change, no violation)
VI-3 MODELS OF ION RETENTION
Using our knowledge of sorption processes we would now like to be able to
predict such phenomena rather than having to explicitly measure it for every condition. A
number of models have been proposed to describe ion retention with varying degrees of
complexity. Computer programs have already been constructed to use many of these
models in assessing the retention of solutes. Accordingly, a brief description of the
models is provided with the main emphasis on the input parameters needed to used
existing programs of these models.
VI-3.1, Chemical Sorption Models
The simplest model is the 'chemical analog' (Kc), which simply considers the reaction as:
sorptive + sorbent <==> sorbate
Kc = (sorbate) / (sorptive)(sorbent)
This is similar in practice to a partition coefficient used for organic or uncharged
molecules. Unfortunately, this model is not very useful for predicting retention processes
because it does not attempt to consider the activity of the sorbate (which we cannot
consider unity like we do other solids). The next two models we will discuss improve
upon this simple equation by considering ion retention on a more molecular basis; they do
not, however, include electrostatic effects. As such they apply to ion retention primarily
on variable charged surfaces: chemical sorption phenomena.
The Freundlich Equation
8
The Freundlich equation was developed empirically, having no theoretical basis,
and is useful for describing the sorption of ions by chemical adsorption and surface
precipitation reactions. Ions of this nature include multivalent cations such as Al3+, Fe3+,
Zn2+, Co2+, and others. In addition, when an ion such as Al or Fe is present in solution,
phosphate may also form a surface precipitate.
x = K•C 1/n
The Freundlich Equation is: m
where,
x = the mass of adsorbate
m = the mass of adsorbent
K = empirical binding coefficient
C = equilibrium adsorptive concentration
n = model dependent factor
In log-log format, giving a linear equation:
x = 1/n log C + log K
log m
Thus, by plotting {log x/m vs log C}, the slope of the resulting line gives [1/n] and the
intercept [log K].
The Langmuir equation
In contrast to the Freundlich equation, the Langmuir equation was developed from
a theoretical standpoint to model the adsorption of gas molecules on surfaces. It was later
applied to the adsorption of ions from solution on mineral surfaces. It works reasonably
well for describing ions that only bind via adsorption mechanisms. Accordingly, most
anions (such as PO43-) conform to the Langmuir equation. One important point to note
about the Langmuir equation is that since it predicts only adsorption phenomena, it only
allows a finite amount of material to be retained on the surface. The maximum amount of
material we can put on a surface is determined by the number of adsorption sites, which
we will term 'monolayer capacity' or 'adsorption maxima'.
Langmuir Equation:
x
K•C•b
m = 1 + K•C
where,
x= the mass of the adsorbate
m= mass of the adsorbent
C= equilibrium adsorptive concentration
K = binding coefficient
b = monolayer capacity of sorbent
9
1
C
In a linear arrangement: xC
m = K b + b
By plotting {C/xm vs C}, the intercept with equal 1/b and the slope 1/bK.
VI-3.2, Models of the Electrostatic Interface
We have consider situations where only chemical forces are modeled or where
only exchange reactions are modeled. Unfortunately, it is not an easy task to consider the
electrostatic surface forces and explicitly incorporate these into a surface model. One of
the more popular theories to describe surface charges is the diffuse layer model. This
model was made popular by the approach of Guoy and Chapman. The Guoy-Chapman
equation assumes that all of the surface charge is satisfied by ions approaching a surface
from solution and forming a "diffuse swarm" of opposite charged ions near the surface.
You should realize that the number of ions in the diffuse swarm is equal to the exchange
capacity of the system. Later electrostatic models incorporated specific planes of
reactivity near the surface with the remaining surface charge being neutralized by a
diffuse swarm, which is again described by the G-C model.
The G-C approach assumes a Boltzmann distribution of ions as a function of the
electrostatic gradient. This leads to an exponential decrease in ion concentration moving
away from the surface, as shown below.
The distribution of cations (a) and charge (b) for a negatively charge surface are
illustrated. The greatest negative charge and cation concentration is at the surface and
exponentially decays to a value found in the bulk solution.
Such a distribution of ions near the surface, a Boltzmann distribution, is described by the
following simple equations.
and,
–(Ze x)
n+(x) = n +() exp
k T
10
(Ze x)
n–(x) = n–() exp
k T
where,
n(x) is the ion concentration at position x and n(∞) in the bulk solution,
z is the ion charge (valence),
is the potential at distance x,
e is the charge on an electron,
k is the Boltzmann constant, and
T is temperature.
The exponential portion of these equation represents the opposing forces of
electrostatic attraction, or repulsion, in the numerator versus thermal random distributions
expressed by the denominator.
Note also that an ion of opposite charge to the surface will be attracted to the
interface, while the ion of the same charge will be repelled. By convention we usually
assume that the surface is negatively charged; but the equations will work whether it is
positively or negatively charged.
All of the terms in the equations above that describe ion distribution in a charged
interface are easily defined except the potential term . Unfortunately, this term is not
measurable and it is rather difficult to calculate. The usual means for obtaining the
potential is using an equation relating the surface charge to the surface potential. The
expression derived in the G-C mode for doing this is:
(Zex)
p = (8RToC•10–3) 0.5sinh (
)
k T
where, p is the total surface charge on the surface.
Fortunately, this equation simplifies at 25°C to:
p = 0.1174 • C∞1/2 sinh( z • 19.46)
Concentration and Charge Effects on the Diffuse-Layer
The G-C model allows us to determine the distribution of charge, potential, and
ion concentrations within the interface. By knowing the ion concentrations, the CEC of
the exchanger (soil) can be determined. More importantly, the G-C model demonstrates
the length to which the surface charge penetrates into the surrounding solution. This
distance is often referred to as the Debye length, which is technically defined as the
distance to which the charge has diminished to a value of 1/e of its charge at the surface.
As you can see from the picture below, both increased 'salt' concentration or an increased
in the ions valence will 'compress' the diffuse layer. This will allow particles to come in
closer proximity to each other and thus promote flocculation of soil particles.
11
Charge neutralization as a function of distance from the surface as
influence by (a) the ionic strength of solution and (b) the valence of the
ions satisfying the charge.
12
EXAMPLE 6-1
By simply measuring the CEC of a soil, you can determined the total charge on a soil
reasonable well. Then by using the G-C model you should be able to calculate the
distribution of ions near the surface.
For example, suppose a soil had a CEC at pH 5 of 50 mmol (+)/Kg and a surface
area of 600 m2/g. Then the surface charge density would be 0.075 Coulombs/m2. (that is
C / m2). With this charge density you can calculate the surface potential using the G-C
expression as follows:
sinh ( * 19.46) =
0.075
= 0.13 V = surface
0.1174 * 0.1
Now, using the Boltzmann expression provided below, you should be able to determine
the concentration of an ion at the surface.
nsurface = nbulk exp{Ze / kT}
e = charge on electron = 1.602 x 10-19 C
k = Boltzmann constant = 1.3805 x 10-23 J / K ( 1 J = V•C)
Z= valence of the ion
T = temperature (K)
n = concentration of the ion
= potential at position x (V)
EXERCISE 6-1
Use the surface potential calculated in EXAMPLE 6-1 for a soil with a CEC of 50
mmol/Kg and a SA=600 m2/g to determine the concentration of:
a) The H+ concentration at the surface if the pH = 5 in the bulk solution
b) Na+ surface concentration if [Na+] = 0.002 M in the bulk solution
c) Al3+ surface concentration if [Al3+] = 1 x 10-5 M in the bulk solution
VI-3.3 Exchange Reactions: Reversible Electrostatic Adsorption
When the adsorption of an ion is dominated by electrostatic forces, especially
those of permanently charged minerals, the resulting complex is a somewhat weak
13
electrostatic interaction. Ions held in this manner are termed 'exchangeable' and their
retention can be best modeled with exchange equations.
The problem with modeling exchange reactions is that we have no way of
obtaining activities of the exchanger complex. The first model proposed for exchange
reactions was developed by Kerr, and is named after him; this model assumes that the
activity of the exchange phase would be comparable to that of the solution.
2 Na-x + Ca2+ = Ca-x + 2 Na+
Kerr type equation:
KKerr = (Ca-X) (Na+)2 / (Na-X)2 (Ca2+)
Unfortunately, this approach works very poorly for soils. Further developments were
made on exchange reactions, however. Two of the more popular models are the
Vanselow and Gapon equations.
Gapon Equation: The Gapon equation was developed empirically and is simple to
calculate.
For the reaction Na-x + 1/2 Ca2+ = Ca1/2-x + Na+
KG =
(Ca 1/2–x) (Na +)
(Ca 2+)1/2 (Na–x)
where KG if the Gapon coefficient.
The Gapon equation works well for describing Na-Ca exchange on smectite and
vermiculite rich soils. This is used extensively to predict Na-Ca exchange in arid
environments. In fact, the U.S. Salinity Laboratory used this approach to develop the
sodium absorption ration (SAR) relationship with the exchangeable sodium ration (ESR).
SAR versus ESR:
[Na–X]
[Na +]
=
K
G
[Ca 1/2–x]
[Ca2+]1/2
[Ca–x][Mg]
assuming Ca and Mg exchange equally, KMg–Ca= 1 = [Mg–x][Ca]
this leads to,
[Na–x]
[Na+ ]
=
K
G
1/2
[Ca 1/2–x] + [Mg1/2–x]
[Ca 2+ + Mg 2+]
14
The US Salinity Lab defined a term SAR{Sodium Absorption Ratio} as
the right side of this equation, with the denominator divided by a factor of
2
SAR = KG
[Na+]
2+
2+
[ Ca +2 Mg ]1/2
And the USSL defined the left side as the Exchangeable Sodium Ratio
(ESR)
[Na–x]
ESR = [Ca –x] + [Mg –x]
1/2
1/2
The also found the EMPIRICAL relationship,
ESR = 0.015(SAR) - 0.01
note: KG = 0.015
This equation was based simply on the fit to a number of soils (SAR vs ESR).
statistically generated equation should be 'calibrated' for specific soils.
Vanselow Equation: This equation was developed in the hope that using mole
factions rather than concentrations might provide an exchange constant or at least a
coefficient applicable over a wide-range of conditions (i.e., it was hoped that mole
fractions would simulate sorbate activities). Unfortunately, neither case prevailed; any
one Vanselow coefficient (Kv) is only valid over a very narrow range of solution
condition. However, as you will see in the next paragraph we can use Kv values to
determine an exchange constant. An example of the Vanselow equation and its
selectivity coefficient is as follows.
For the reaction,
2 Na-x + Ca2+ = Ca-x + 2 Na+
Kv =
NCa [Na+] 2
N2Na [Ca 2+]
where N is the mole fraction, e.g., NCa =
QCa–x
, and Q is the
QCa–x + Q Na–x
number of moles of the ion on the exchanger (-x).
Such a
15
Neither the Gapon or the Vanselow coefficients are true equilibrium constants. To obtain
a true exchange reaction constant we must use the Vanselow equation and consider all
possible solution conditions for the given ions. This means that we must move from
having coefficients for exclusively one ion on the surface to coefficients for solely the
other ion on the surface; we must also have data for all conditions in between. Then we
can integrate the coefficients over these conditions and obtain the true exchange constant.
Exchange constant:
1
ln Kex =
0
ln Kv dNB
VI-3.4, Surface Complexation Models
The surface complexation models attempt to incorporate both chemical and
electrostatic factors in the description of ion retention. In doing so, they are based on a
microscopic interpretation of the surface. The two dominant models are the Stern Model
(which is often now just called the double-layer model) and the triple-layer model. The
Figure below presents a schematic illustration of these models. Both of these models use
the diffuse-layer theory to represent the outer portion of ions attracted to a surface. As we
move closer to the surface these models attempt to address chemical interactions by
allowing some ions to enter a plane very near the surface. They also account for the
energies needed for an ion to pass through the electrostatic gradient.
In the Stern (2-Layer) model, a layer of ions closely associated with the surface
are predicted for form; this is termed the 'Stern layer'. In contrast to the exponential decay
of charge in the diffuse layer, the Stern layer has a linear decline in charge with distance.
The rate of decline, or the slope of the charge versus distance curve, depends on the
capacitance (C1) of this layer, which is dependent on the total charge and type of ions in
this layer. Beyond the Stern layer, the charge decays as described by the Guoy-Chapman
diffuse layer equation.
The Triple-layer model is an extension of the Stern model; it provides a third
plane to accommodate inner-sphere complexes, outer-sphere complexes, and the diffuse
swarm. Inner-sphere complexes reside in the layer closest to the surface (the layer),
outer-sphere complexes are present in the next plane (the layer), and the requisite
surface charge is neutralized by ions in the diffuse layer. Like the Stern layer, the charge
decreases linearly in both the (with a capacitance of C1) and (with a capacitance of
C2) layers.
The charges of the Stern, , and layers are expressed:
Stern or layer: = C1
layer: = C2
16
and the charge in the diffuse layer is represented by the G-C model we described earlier.
To finish our modeling, we must express the adsorption reactions for each layer. This is
simply a mass action balance equation representing ions that would occupy each layer,
sorptive + sorbent <==> sorbate
K = (sorbate) / (sorptive) (sorbent)
so that there would be three equilibrium coefficients, one for each layer. The computer
code will then not only account for these reactions but it will also factor in the effects of
an electrostatic gradient on the ability of the sorptive to become a sorbate.
Surface complexation models are becoming increasing more popular and useful as
the parameters for different systems are defined. Additionally, many computer codes now
have surface complexation models built into their chemical speciation routines. For
further information on this subject, an excellent reference is : Surface Complexation
Modeling by Dzomback and Morel, Wiley and Sons Publishing, NY. 1990.
17
Stern Model
Surface
Stern
Layer
Diffuse Layer
Triple-Layer Model
-Layer
-Layer
Surface
Diffuse Layer
-
O
O Al
Fe
O
O Zn
Fe
C1
p
Cu
C1
-
p
distance
C2
-
distance
The Stern and Triple-layer models address both electrostatic and chemical
factors in ion retention. The Stern model allows one plane of closest approach
while the Triple-layer has two, one for inner-sphere complexes and one for
outer-sphere complexes. Both models use the G-C diffuse layer expression to
model the charge and ion distributions beyond these inner adsorption planes.
For Details see: Dzomback and Morel. 1990. Surface Complexation Modeling. Wiley
and Sons.
18
VI-4 CHOOSING A MODEL
A Flow chart summarizing the choice of models is provided below. By using is chart
you should be able to use the simplest model for any specific condition.
MODEL FLOW-CHART
Chemically Reactive
Surface & Ions
no
yes
Exchange
Equations
Surface Precipitation
Probable
partial
yes
Freundlich
Equation
Na-Ca
exchange on
smectite
no
Langmuir
Equation
yes
Gapon
Equation
Surface Complexation
Models
no
Vanselow
Equation