Supplemental Chemistry Experimental Procedures
advertisement

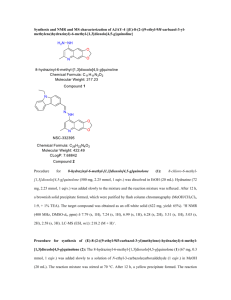
Supplemental Text File 1. Chemistry Experimental Procedures All chemicals and solvents were obtained from Sigma-Aldrich (Milwaukee, WI) of Fisher Scientific (Pittsburg, PA) and used without further purification. Analytical HPLC was performed on a Varian Prostar system, with a Varian Microsorb-MW C18 column (250 x 4.6 mm; 5 ) using the following solvent system A= H2O/0.l % TFA and B=acetonitrile/0.1 % TFA. Varian Prepstar preparative system equipped with a Prep Microsorb-MW C18 column (250 x 41.4 mm; 6 ; 60 Å) was used for preparative HPLC with the same solvent systems. Program A: Gradient: 0-5 min. 30 % B. 5-35min. 95 % B. 30-35 min. 95% B. Program B: Gradient: 0-5 min. 10 % B. 5-35min. 95% B. 30-40 min. 95% B. Mass spectra (ionspray, a variation of electrospray, unless otherwise noted) were acquired on an Applied Biosystems Q-trap 2000 LC-MS-MS. UV was measured on a Perkin Elmer Lambda 25 UV/Vis spectrometer. IR was measured on a Perkin Elmer Spectra One FT-IR spectrometer. 1 H-NMR and 13 C-NMR spectra were recorded on a Brucker Biospin spectrometer with a B-ACS 60 autosampler: 600.13 MHz for 1H-NMR and 150.92 MHz for 13 C-NMR. Chemical shifts (δ) are determined relative to d4- methanol (referenced to 3.34 ppm (δ) for 1H-NMR and 49.86 ppm for l3 C-NMR). Proton-proton coupling constants (J) are given in Hertz and spectral splitting patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet or overlapped (m), and broad (br). Flash chromatography was performed using Merck silica gel 60 (mesh size 230-400 ASTM) or using an Isco (Lincoln, NE) combiFlash Companion or SQ16x flash chromatography system with RediSep columns (normal phase silica gel (mesh size 230-400ASTM)) and Fisher Optima TM grade solvents. Thin-layer chromatography (TLC) was performed on Merck (Darmstadt, Germany) silica gel F-254 aluminum-backed plates with visualization under UV (254nm) and by staining with potassium permanganate or ceric ammonium molybdate. Chemical synthesis of ON012380. ON012380 was synthesized following the general procedure described in patent WO 03/0720621 (supplement figure 1). Synthesis of 4-Methoxv-3-Nitro benzylbromide(1) : A solution of 4-methyl-2-nitroanisole (10g, 60 mmol), N-bromosuccinimide (10.6 g, 60 mmol) and benzoyl peroxide (14.5 g, 60 mmol) in carbontetrachloride (240 mL) was heated at reflux for 18h. The reaction mixture was then poured into water and solid separated was filtered. The aqueous layer was extracted with carbontetrachloride (3 x 100 mL) and the organic phase was separated and evaporated to give a solid product. The solid products were combined and recrystallized from ethyl acetate-hexane to give a crystalline product of 3-nitro-4-methoxy benzyl bromide. m.p.110-112 oC, yield 70-75%. 1 H NMR (DMSO-d6) 8.02 (1H, d, J = 1.8 Hz), 7.77 (1H, dd, J = 9, 1.8 Hz), 7.38 (1H, d, J = 8.4 Hz), 4.76 (2H, s), 3.95 (3H, s). 13 C NMR (DMSO-d6) 151.81, 138.81, 135.41, 130.53, 125.57, 114.70, 56.91, 32.65. Synthesis of 4-Methoxv-3-Nitrobenzylthioacetic acid (2): To a cold solution of sodium hydroxide (2.7 g, 81 mmol) in methanol (80 mL), thioglycollic acid (3 g, 40 mmol) was added slowly over 30 minutes. Sodium thioglycollate precipitated was dissolved by stirring and warming up the solution. The solution was cooled to room temperature and 4-methoxy-3- nitrobenzyl chloride (8 g, 40 mmol) was added in portions to reduce the intensity of exothermic reaction. The reaction mixture was then refluxed for 4 hours, cooled and poured onto crushed ice (1 Kg) containing hydrochloric acid (11 mL). The precipitate formed was filtered, washed with ice cold water and dried under vacuum. (8 g, 95 % yield) m.p.130-132 oC. . MS 275.2 (M+NH4). Synthesis of 4-Methoxy-3-Nitrobenzylsulfonylacetic acid (3): 4-methoxy-benzylthioacetic acid (8 g, 31 mmol) was dissolved in glacial acetic acid (65 mL) and 30 % hydrogen peroxide (16 mL) was added in one portion and the mixture was stirred at room temperature for 10 hours. The contents of the flask were cooled and poured on to the crushed ice (400 g). The yellow precipitate formed, filtered, washed with cold water and dried (55 % yield). Recrystallization from hot water yielded crystals 4-methoxy-3-nitrobenzylsulfonyl acetic acid. m. p. 96-98 oC. MS 307.2 (M+NH4). 1 H NMR (DMSO-d6) 7.93 (1H, d, J = 2.0 Hz), 7.69 (1H, dd, J = 8.4, 1.8 Hz), 7.43 (1H, d, J = 7.8 Hz), 4.69 (2H, s), 4.23 (2H,s), 3.90 (2H,s), 3.93 (2H, s), 3.84 (3H, s), 13C NMR (DMSO-d6) 164.45, 152.39, 138.78, 137.29, 127.48, 120.11, 114.60, 57.28, 56.57, 55.52. Synthesis of (E)-2,4,6-Trimethoxvstyrvl-4-Methoxy-3-Nitrobenzylsulfone (4): A solution of 4-methoxy-3-nitrobenzylsulfonylacetic acid (7g, 24.2 mmol) in 50 mL of glacial acetic acid was treated with 2,4,6-trimethoxybenzaldehyde (4.84 g, 24.2 mmol) in the presence of catalytic amounts of benzylamine (1 mL). The reaction mixture was refluxed for 6 hours and acetic acid was removed under vacuum. The gummy material obtained was treated with 2-propanol to yield a solid product which was recrystallized from a mixture of acetic acid and 2propanol. Yield 28%, m. p.186-187 oC. MS 424.2 (M+H). 1 H NMR (DMSO-d6) 7.89(1H, d, J = 2.0 Hz), 7.65 (1H, dd, J = 8.4, 1.8 Hz), 7.52 (1H,d, J = 15Hz), 7.52 (1H,d, J = 15 Hz), 7.38(1H, d, J = 8.4Hz), 7.10(1H, d, J = 15 Hz), 6.29(s,1H), 4.60 (2H, s), 3.93 (3H,s), 3.85 (9H, s), 13C NMR (DMSO-d6) 163.80, 160.96, 152.04, 138.56, 137.13, 134.01, 127.32, 122.66, 121.82, 114.25, 102.37, 90.88, 58.27, 56.72, 56.01, 55.60. Synthesis of (E)-2, 46-Trimethoxvstvrvl-4-Methoxy-3-Nitrobenzylsulfone (5): A solution of (E)-2, 4, 6-trimethoxystyryl-4-methoxy-3-nitrobenzylsulfone (4) (2 g, 4.72 mmol) (4) in acetone-water 60 ml (10:5) was heated to 50oC. After 30 min, sodium hydrosulfite (Na2S204) (19.3g, 95 mmol) was added slowly, and the mixture was heated at reflux (50 C,1 h. ), cooled to room temperature and water was added. The product was rinsed withNaHC03, and then isolated by extraction with ethyl acetate. The organic layer was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was recrystallized from 2propanol, m. p.148-150 oC. . MS 394.3 (M+H). 1 H NMR (DMSO-d6) 7.58 ( 1H, d, J = 15 Hz), 7.08 (1H, J = 15 Hz), 6.75 (1H,d, J = 8Hz), 6.63 (1H, d, J = 2Hz), 6.50 (1H,dd, J =7.8,2 Hz), 6.29(s,1H), 4.84 (2H, br), 4.21 (s, 2H), 3.84 (9H,s), 3.82 (3H, s). Synthesis of (E)-2, 4, 6-Trimethoxystyryl-3-amino-substituted-4- methoxvbenzyrlsulfone derivatives (6): To a stirred solution of methyl bromoacetate (0.7 ml, 6.35 mmol) and sodium acetate (0.53 g, 6.35 mmol) in methanol (20 mL) was added 2,4, 6-trimethoxy-3-amino-4methoxy- benzysulfone (5) (0.5 g, 1.27 mmol) and stirring was continued under reflux temperature for 12- 15 h. The contents of the flask were cooled and poured onto the ice. The resultant ester obtained (85 % yield) was filtered (m. p. 182-185°C). MS 480.3 (M+H). Crystal structure was obtained for the compound 6. The compound is racemic around C20A. (Fig. 2). 1 H NMR (DMSO-d6) 7.55(1H, d, J = 15 Hz), 7.09 (1H, dd=, J = 15 Hz), 6.80 (1H,d, J = 8.4Hz), 6.60 (1H,dd, J =8.4, 1.8 Hz), 6.44(1H, d, J = 1.8 Hz), 6.29(2H, s), 4.98 (1H, d, J =7.4 Hz), 4.27 (2H, s), 4.02 (1H, br), 3.85 (9H, s), 3.84 (3H,s), 3.61 (3H, s), 1.35 (3H, d, J = 7.0 Hz). 13 C NMR (DMSO-d6) 174.28, 163.57, 160.89, 146.44, 135.92, 132.73, 125.58, 121.31, 119.67, 112.38, 109.58, 102.53, 90.87, 60.26, 55.99, 55.59, 55.39, 51.87, 50.66, 18.1. Synthesis of ON012380 2: A solution of ester (6) (100 mg, 0.2 mmol) in 10 ml DCM was added NaOSiMe3 (34 mg, 0.3 mmol) and the mixture was stirred at r.t. overnight. Remove of the solvent and added 1N HCl aqueous solution and the mixture was extracted with DCM. The organic layer was separated, washed with water and dried over anhydrous sodium sulfate. Volatiles were removed under vacuum and the product. MS 466.3 (M+H). 1H NMR (DMSO-d6) 7.55 (1H, d, J = 15 Hz), 7.11 (1H, d, J = 15 Hz), 6.80 (1H, d, J = 7.8 Hz), 6.59 (1H, dd, J = 7.8, 1.8 Hz), 6.48 (1H, d, J = 1.8 Hz), 6.29 (2H, s), 4.27 (2H,s), 3.90 (1H,q, J = 7.2 Hz), 3.85 (6H, s), 3.84 (3H, s), 3.79 (3H, s), 1.31 (3H, d, J = 7.2 Hz). C NMR 13 175.19, 172.05, 163.6, 160.9, 146.4, 135.99, 132.76, 123.64, 121.38, 119.41, 112.33, 109.57, 102.58, 90.93, 60.33, 56.04,55.63,55.45, 50.47, 21.07, 18.29. Reference: 1. PREMKUMAR, R.; RAMANA, R.; STANLEY, B. AMINO-SUBSTITUTED (E)-2. 6-DIALKOXYSTYRYL 4-SUBSTITUTEDBENZYLSULFONES FOR TREATING PROLIFERATIVE DISORDERS WO03072062 2003-09-04 2003. 2. Laganis, E. D.; Chenard, B. L., Metal Silanolates: Organic Soluble Equivalents for O-2. Tetrahedron Letters 1984, 25, (51), 5831-5834. Experimental Data Collection A colorless plate crystal of C23H29NO8S having approximate dimensions of 0.44 x 0.25 x 0.10 mm was mounted on a Mylar loop. All measurements were made on a Rigaku RAXIS SPIDER imaging plate area detector with graphite monochromated CuK radiation. Indexing was performed from 4 oscillations that were exposed for 60 seconds. The crystal-to-detector distance was 127.40 mm. Cell constants and an orientation matrix for data collection corresponded to a primitive triclinic cell with dimensions: a = b = c = V = 7.5873(8) Å 9.5354(10) Å 17.614(2) Å 1155.5(2) Å3 = 97.696(8)o = 96.712(8)o = 111.541(7)o For Z = 2 and F.W. = 479.54, the calculated density is 1.378 g/cm3. Based on a statistical analysis of intensity distribution, and the successful solution and refinement of the structure, the space group was determined to be: P-1 (#2) The data were collected at a temperature of -100 + 1oC to a maximum 2 value of 136.4o. A total of 136 oscillation images were collected. A sweep of data was done using scans from 20.0 to 200.0o in 5.0o step, at =0.0o and = 0.0o. The exposure rate was 36.0 [sec./o]. A second sweep was performed using scans from 30.0 to 120.0o in 5.0o step, at =54.0o and = 0.0o. The exposure rate was 36.0 [sec./o]. Another sweep was performed using scans from 30.0 to 100.0o in 5.0o step, at =54.0o and = 90.0o. The exposure rate was 36.0 [sec./o]. Another sweep was performed using scans from 20.0 to 180.0o in 5.0o step, at =54.0o and = 180.0o. The exposure rate was 36.0 [sec./o]. Another sweep was performed using scans from 20.0 to 200.0o in 5.0o step, at =54.0o and = 270.0o. The exposure rate was 36.0 [sec./o]. The crystal-to-detector distance was 127.40 mm. Readout was performed in the 0.100 mm pixel mode. Data Reduction Of the 5103 reflections that were collected, 921 were unique (Rint = 0.027); equivalent reflections were merged. The linear absorption coefficient, , for Cu-K radiation is 16.740 cm-1. An empirical absorption correction was applied which resulted in transmission factors ranging from 0.682 to 0.846. The data were corrected for Lorentz and polarization effects. Structure Solution and Refinement The structure was solved by direct methods1 and expanded using Fourier techniques2. Most non-hydrogen atoms were refined anisotropically; one pair of disordered carbon atoms was refined isotropically. Hydrogen atoms were used in calculated positions, except for the amine H atom, which was refined. The final cycle of full-matrix least-squares refinement3 on F2 was based on 3857 observed reflections and 306 variable parameters and converged (largest parameter shift was 0.01 times its esd) with unweighted and weighted agreement factors of: R1 = ||Fo| - |Fc|| / |Fo| = 0.0367 wR2 = [ ( w (Fo2 - Fc2)2 )/ w(Fo2)2]1/2 = 0.0962 The standard deviation of an observation of unit weight4 was 1.13. Unit weights were used. The maximum and minimum peaks on the final difference Fourier map corresponded to 0.27 and -0.32 e-/Å3, respectively. Neutral atom scattering factors were taken from Cromer and Waber5. Anomalous dispersion effects were included in Fcalc6; the values for f' and f" were those of Creagh and McAuley7. The values for the mass attenuation coefficients are those of Creagh and Hubbell8. All calculations were performed using the CrystalStructure9,10 crystallographic software package except for refinement, which was performed using SHELXL-9711. The structure is racemic; both enantiomers exist in the unit cell, related by the inversion center in space group #2 (P-1). In addition, there is a slight disorder at each site such that the enantiomers are disordered. In the final model, atoms C20 and C21 are disordered 85.8% in the major orientation (and therefore 14.2% in the minor orientation). The existence of the racemic mixture was verified by solving the structure in the chiral space group P1 (#1). In space group P1, there are two independent molecules in the asymmetric unit, and they represent both enantiomers. If the substance was enantiomerically pure, both of the independent molecules in space group P1 would be the same enantiomer. References (1) SIR92: Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M., Polidori, G., and Camalli, M. (1994) J. Appl. Cryst., 27, 435. (2) DIRDIF99: Beurskens, P.T., Admiraal, G., Beurskens, G., Bosman, W.P., de Gelder, R., Israel, R. and Smits, J.M.M.(1999). The DIRDIF-99 program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, The Netherlands. (3) Least Squares function minimized: (SHELXL97) w(Fo2-Fc2)2 where w = Least Squares weights. (4) Standard deviation of an observation of unit weight: [w(Fo2-Fc2)2/(No-Nv)]1/2 where: No = number of observations Nv = number of variables (5) Cromer, D. T. & Waber, J. T.; "International Tables for X-ray Crystallography", Vol. IV, The Kynoch Press, Birmingham, England, Table 2.2 A (1974). (6) Ibers, J. A. & Hamilton, W. C.; Acta Crystallogr., 17, 781 (1964). (7) Creagh, D. C. & McAuley, W.J .; "International Tables for Crystallography", Vol C, (A.J.C. Wilson, ed.), Kluwer Academic Publishers, Boston, Table 4.2.6.8, pages 219-222 (1992). (8) Creagh, D. C. & Hubbell, J.H..; "International Tables for Crystallography", Vol C, (A.J.C. Wilson, ed.), Kluwer Academic Publishers, Boston, Table 4.2.4.3, pages 200-206 (1992). (9) CrystalStructure 3.7.0: Crystal Structure Analysis Package, Rigaku and Rigaku/MSC (2000-2005). 9009 New Trails Dr. The Woodlands TX 77381 USA. (10) CRYSTALS Issue 10: Watkin, D.J., Prout, C.K. Carruthers, J.R. & Betteridge, P.W. Chemical Crystallography Laboratory, Oxford, UK. (1996) (11) SHELX97: Sheldrick, G.M. (1997). EXPERIMENTAL DETAILS A. Crystal Data (Supplement Figure 2) Empirical Formula C23H29NO8S Formula Weight 479.54 Crystal Color, Habit colorless, plate Crystal Dimensions 0.44 X 0.25 X 0.10 mm Crystal System triclinic Lattice Type Primitive Indexing Images 4 oscillations @ 60.0 seconds Detector Position 127.40 mm Pixel Size 0.100 mm Lattice Parameters a = 7.5873(8) Å b = 9.5354(10) Å c = 17.614(2) Å = 97.696(8) o = 96.712(8) o = 111.541(7) o V = 1155.5(2) Å3 Space Group P-1 (#2) Z value 2 Dcalc 1.378 g/cm3 F000 508.00 (CuK) 16.740 cm-1 B. Intensity Measurements Diffractometer Rigaku RAXIS-RAPID Radiation CuK ( = 1.54180 Å) graphite monochromated Detector Aperture 280 mm x 256 mm Data Images 136 exposures Exposure Rate 36.0 sec./o oscillation Range (=0.0, =0.0) 20.0 - 200.0o oscillation Range (=54.0, =0.0) 30.0 - 120.0o oscillation Range (=54.0, =90.0) 30.0 - 100.0o oscillation Range (=54.0, =180.0) 20.0 - 180.0o oscillation Range (=54.0, =270.0) 20.0 - 200.0o Detector Position 127.40 mm Pixel Size 0.100 mm 2max 136.4o No. of Reflections Measured Total: 5103 Corrections Unique: 10146 (Rint = 0.027) Lorentz-polarization Absorption (trans. factors: 0.682 - 0.846) C. Structure Solution and Refinement Structure Solution Direct Methods (SIR92) Refinement Full-matrix least-squares on F2 Function Minimized w (Fo2 - Fc2)2 Least Squares Weights w = 1/ [ 2(Fo2) + (0.0430 . P)2 + 0.2438 . P ] where P = (Max(Fo2,0) + 2Fc2)/3 2max cutoff 136.4o Anomalous Dispersion All non-hydrogen atoms No. Observations (All reflections) 3857 No. Variables 306 Reflection/Parameter Ratio 12.60 Residuals: R1 (I>2.00(I)) 0.0367 Residuals: R (All reflections) 0.0483 Residuals: wR2 (All reflections) 0.0962 Goodness of Fit Indicator 1.126 Max Shift/Error in Final Cycle 0.011 Maximum peak in Final Diff. Map 0.27 e-/Å3 Minimum peak in Final Diff. Map -0.32 e-/Å3





![Synthesis of [ChCl][ZnCl2]2 ionic liquid 1 mmol of choline chloride](http://s3.studylib.net/store/data/006941233_1-367e4dc6f52fe7a83ef718561a45342d-300x300.png)




