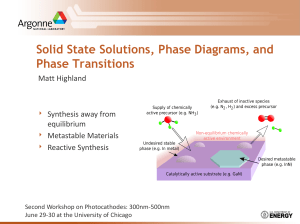
During the last two decades novel materials have been widely
advertisement

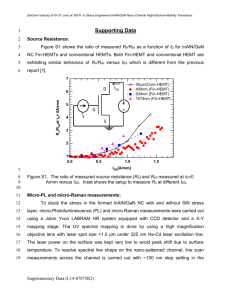
World Journal Of Engineering AB INITIO STUDY OF STRUCTURAL AND ELECTRONIC PROPERTIES OF INN/SI AND GAN/SI INTERFACES Mutigullin I.V.1, Abgaryan K.K.1, Bazhanov D.I.2, Stepanyuk O.V.1 1 Institution of Russian Academy of Sciences Dorodnicyn Computing Centre of RAS, Vavilov st. 40, 119333 Moscow, Russia. 2 Faculty of Physics M.V. Lomonosov Moscow State University, Leninskie Gory, Moscow, Russia. Introduction One of the main priorities during the development of civil and military innovative radioelectronic defense equipment is the production of the new semiconductor film nanostructures based on nitride or oxide compounds bearing unique physical and mechanical properties required for solid state microwave electronics. In this context very promising are such nitride semiconductors as GaN, InN, AlN etc. One of the most attractive substrates for epitaxial nitride growth is Si [1]. But due to a large lattice mismatch between Si(111) substrates and nitride (0001) surface the interface is characterized by large strain [2, 3]. In this work we present theoretical investigation of adhesion properties of InN(0001)/Si(111) and GaN(0001)/Si(111) interfaces as well as influence of strain on band-gap value. Theory All the calculations presented in this article were based on Density functional theory (DFT) within the local-density approximation LDA scheme widely used to study the ground state properties of solids and their surfaces. [4, 5] DFT LDA simulations were performed using the Vienna ab initio simulation package VASP based on pseudopotentials and a plane-wave basis set to solve the Kohn–Sham equations with periodic boundary conditions. [6] To study the properties of InN/Si and GaN/Si interfaces, we used a common slab approximation with a supercell containing 21 atomic layers and a large vacuum space. All the atomic layers in the slabs were allowed to relax. The relaxation process was stopped when the residual force acting on each atom in the slab was less than 0.01 eV/Å. Ef InN DOS, N states 20 15 10 5 0 -20 -15 -10 -5 0 5 10 15 Energy (eV) Ef GaN DOS, N states 20 15 10 5 0 -20 -15 -10 -5 0 5 10 15 Energy (eV) Fig. 1. Calculated density of states for bulk InN and GaN. Results and Discussion Both GaN and InN crystallize predominantly in wurtzite structure [1]. Calculated bulk lattice constants are in a good agreement with experimental data [1]. For GaN obtained lattice constant a=3,15 Å and c/a=1.63 while for InN a=4.50 Å and c/a=1.62. It is well known that LDA due to the so-called “band-gap problem” underestimates the value of bad-gap of semiconductors [7]. Thus we used LDA+U [8] to calculate electronic properties of GaN and InN. In Fig. 1 we demonstrate obtained 835 World Journal Of Engineering electronic densities of states for GaN and InN. Calculated band-gaps are 3.45 eV and 0.79 eV for GaN and InN respectively, while experimental values are 3.5 eV [9] and 0.65-0.7 eV [10] respectively. GaN respectively. In the case of coherent InN(0001)/Si(111) interface the lattice mismatch is +9% and our calculations revealed a strong band-gap decreasing till 0.12 eV. InN(0001)/Si(111) coherent interface with Si lattice constant (5,40 Å) was studied as an example of the formation of semiconductorsubstrate interface. Different possible termination configurations were considered. The most stable interface geometry appears to be when N surface-terminating atom is located above surface Si atom. Calculated adhesion energies are presented in Table 1. The strongest adhesion corresponds for the case of N-termination with terminating Natom on top of surface Si atom. Table 2. Calculated InN and GaN bandgap in dependence from relative lattice constant change. -2% -1% 0% +1% +2% Equilibrium value InN 0,93 0,85 0,79 0,69 0,56 GaN 3,73 3,57 3,45 3,27 3,08 Conclusion First-principles studies have been performed for InN(0001)/Si(111) interface. Adhesion energies were calculated for different positions of atoms at interface. The influence of strain driven lattice constant change on InN and GaN band-gap was studied. References 1. X. Wang, A. Yoshikawa, Progress in Crystal Growth and Characterization of Materials 48/49 (2004) 42-103 2. T. A. Rawdanowicz, J. Narayan, Appl. Phys. Let. 85 (2004) 133. 3. A.G. Bhuiyan, A. Hashimoto, A. Yamamoto, J. Appl. Phys. 94 (2003) 2779 4. P. Hohenberg, W. Kohn, Phys. Rev. 136 (1964) B864. 5. W. Kohn, L. J. Sham, Phys. Rev. 140 (1965) A1133. 6. G.Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 11169. 7. J.P. Perdew, M. Levy, Phys. Rev. Let. 51 (1983) 1884. 8. V. I. Anisimov, F. Aryasetiawan, A. I. Lichtenstein, J. Phys.: Condens. Matter 9 (1997) 767. 9. K. Lawniczak-Jablonska, T. Suski, I. Gorczyca et al., Phys. Rev. B 61 (2000) 16623. 10. V.Yu. Davydov, A.A. Klyuchihin, Fizika i tehnika poluprovodnikov 38 (2004) 897. This work has been financed through RFBR: grants #10-08-01263-a and #09-01-13541ofi-c. Table 1. Adhesion energy for different configurations of InN(0001)/Si(111) interface ((0;0) – coordinate corresponds to Si surface atom, (2/3;1/3) – next substrate level). Termination Coordinate of Adhesion type terminating energy, atom (In or N) J/м2 N (1/2;0) -0,95 N (0;0) -2,55 N (1/3;2/3) -1,01 N (2/3;1/3) -0,59 In (1/2;0) -1,69 In (0;0) -1,61 In (1/3;2/3) -1,67 In (2/3;1/3) -1,57 Since Si(111) and InN equilibrium lattice constants (3,82 Å and 3.15 Å respectively) are different, interface formation is followed by significant stress distribution. Lattice mismatch effects strongly the electronic properties of nitride film, particularly the band-gap. Thus we performed calculations of band-gap dependence on the lattice constant. Generally, our calculations reveal a strong influence of the lattice constant on band-gap value (see Table 2). In particular, the decreasing of lattice constant up to 2% led to the bandgap broadening up to 17% for InN and 8% for GaN, while the increasing of lattice constant up to 2% resulted in 29% and 13% narrowing of bandgap for InN and 836