KcsA_paper_15_text - McDermott
advertisement

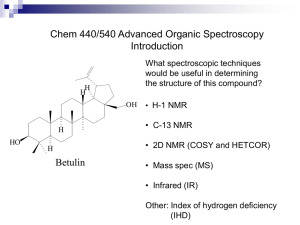
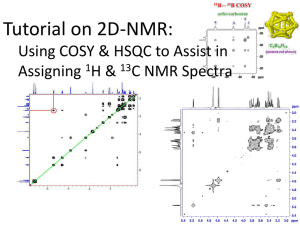
1 Solid State NMR Study and Assignments of the KcsA Potassium Ion Channel of S. lividans Krisztina Varga[a], Lin Tian[a], Joshua A. Wand[b], Ann E. McDermott[a]* Contributed from the Department of Chemistry, Columbia University, New York, NY 10027, Department of Biochemistry and Biophysics, University of Pennsylvania, Philadelphia, PA, 19104 * To whom correspondence should be addressed: Columbia University, Department of Chemistry, 3000 Broadway, New York, NY 10027, Phone: (212) 854-xxxx, email: aem5@columbia.edu [a] Columbia University [b] University of Pennsylvania Abstract: We carried out solid state NMR (SSNMR) studies leading site-specific assignments of uniformly 13C, 15N enriched microcrystalline KcsA potassium channel (160 residues) from Streptomyces lividans, an integral membrane protein. The experimental strategy included a combination of 2D and 3D solid state NMR techniques at 600 and 750 MHz. For most amino acids, signal patterns were established in 2D 13C13 C experiments. Since the secondary structure is non-helical in this region of the protein, the chemical shifts of some residues are shifted from the typical helical residues and provide a useful fingerprint for the sample. In the 3D spectra, residue types were confirmed using side chain assignments. Assignments include both backbone and some side chains atoms near and at the selectivity filter region of the protein. Potassium channels are widely studied for their very efficient conductivity and selectivity, and our study opens the door for structure determination and mechanism of ion conduction by SSNMR. Introduction The purpose of this work was to establish the feasibility of site-specific assignments and further biophysical studies of KcsA potassium ion channel from Streptomyces lividans (S. lividans), a 160 amino acid integral membrane protein. Potassium channels are fascinating proteins present in both prokaryotes and eukaryotes. Two remarkable features of these channels are the very high selectivity towards K+ ions and their rates of conductivity which approach the diffusion limit.1 KcsA functions as a homotetramer (70.4 kDa). The monomer is composed of two trans-membrane helices connected by an extracellular loop including the pore helix. The pore region sequence is very conserved, thus likely all K+ channels (both ligand- and voltage-gated) from different species essentially have the same gating mechanism and pore structure 1-3. KcsA, isolated from S. lividans, is an integral membrane protein with similar structure and ion permeability to other K+ channels. It is the first K+ channel for which the 3D structure has been determined by X-ray diffraction to 3.2 Å resolution1 by MacKinnon and colleagues. 2 Later the resolution was refined to 2.0 Å for the channel-Fab complex.4 The crystals used for both X-ray studies (PDB: 1BL81 and 1K4C4) were grown of a deletion version of KcsA – both the N and C terminals were deleted. The availability of X-ray structural information can facilitate NMR structural studies and makes KcsA a good model system for other membrane protein studies. Conversely, solid state NMR can offers advantages over other biophysical methods. One of the advantages of solid state NMR studies is that X-ray quality crystals are not required – microcrystalline protein precipitates are suitable for obtaining high resolution spectra.5 Thus it is feasible to study the functional, whole channel under various conditions, including ligands, inhibitors, and toxins bound to it. NMR chemical shift changes report on structural changes which may lead to a better understanding of the workings of postassium channels. In this study KcsA was precipitated in detergent micelles, however in subsequent studies the enzyme can be reconstituted into lipid bicelles, vesicles or as 2D crystals, which mimic the natural membrane environment even more closely. Solid state NMR also offers the possibility to study protein dynamics of various resolved sites. The prerequisite of structural studies of any protein by NMR is the assignment of resonance peaks – the identification of which resonance peak corresponds to which amino acid site specifically. KcsA is an integral membrane protein which exists mostly in alpha-helical conformation (~50%), however other secondary structural elements are also present6. There is high correlation between protein secondary structure and chemical shifts 7-9. It is very difficult to assign all alpha helical proteins by NMR due to spectral overlap. Beta sheet and other types of secondary structure elements facilitate assignments because the chemical shift of those amino acids will be resolved from the alpha-helical shifts. In KcsA, the functionally most interesting region, the pore (containing the selectivity filter) is of mixed secondary elements thus was expected to be most easily assignable. All K+ channels contain a critical, highly conserved amino acid ‘signature sequence’ 10, 11, which makes the selectivity filter of the channels and is essential for proper function The signature sequence consists of the following amino acids: T75, V76, G77, Y78, and G79 (in KcsA). To create a stack of main chain carbonyl oxygen rings ligating the K+ ion, which is the structural basis of the channel selectivity, the conformation of the five residues composing the selectivity filter alternates between left- and right-handed alpha-helix, L and R respectively. The unique conformation results in unusual backbone torsion angles, which yielded unusual shifts in the NMR spectra for some of these residues. For instance, there were two valines easily distinguishable from the other fourteen valine residues, which are likely part of the nonhelical pore region (Figure 2). Understanding the three-dimensional structures and mechanisms of proteins becomes increasingly important since it advances all the other areas of life sciences, such as biology, pharmacology, and medicine. In the last few decades, the two major tools of structural biology have been solution NMR and X-ray crystallography, and by the end of 2005 more than 30,000 soluble protein structures were solved to atomic resolution and deposited in the Protein Data Bank (PDB), a web based single worldwide repository of 3D biological macromolecular structure data12. In contrast, integral membrane proteins still pose a considerable challenge to biophysicists. Even though they constitute 20-30% of the genome, only a handful membrane protein structures were determined (<2% of known structures13) by these methods due to their low solubility and difficult 3 crystallization. Integral membrane proteins are involved in essential cell functions, such as signal detection, import and export of nutrients and ions, cell-to-cell communication, and energy production. Membrane proteins are a major part of drug receptors and a better understanding of their structure and workings would lead to a significant improvement in drug design and development. Recent technical advances in solid state NMR (ssNMR) make it a very promising method for exploring membrane proteins and other non-soluble or large biological structures that are difficult to study by solution NMR and X-ray crystallography. Once the site specific assignments are obtained, the sufficient number of structural constraints must be measured for 3D protein structure determination. In the last few years, a large number of magic angle spinning (MAS) multidimensional techniques developed at high fields which facilitated the assignment of solid protein samples. Probably the first study that yielded spectra of high enough resolution for site-specific assignments was of microcrystalline BPTI in 200014. Since then, a few uniformly or extensively 13C, 15N enriched ssNMR soluble protein assignments were published15-24. Membrane proteins present a bigger challenge than soluble proteins, and to date only the LH2 light harvesting complex25, 26 has nearly complete assignments. There are other membrane protein assignments in progress by various research groups: Outer-membrane protein G27, Diaglycerol kinase28, and the LH1 light-harvesting complex29. The broader impact of the development of assignments techniques in the solid state is that it would open new avenues to study protein structures and dynamics. To date, a few small peptide30, 31 and protein (i.e. ubiquitin32, 33, -spectrin SH3 domain34, and kaliotoxin35) 3D structures have been determined by solid state NMR. KcsA is a well-suited sample for ssNMR studies was chosen as a model system for several reasons: stability, availability, and biological importance. Long life time and thermal stability make KcsA a very favorable target for ssNMR studies, since with the currently available techniques the minimum experiment time is approximately a month for collecting the 2D and 3D spectra necessary for assignment work. Sample heating is another general consideration in MAS ssNMR experiments – samples heat up because of the frictional forces due to spinning at high speed (~ 10-13 kHz). Even more importantly, hydrated, high salt containing protein samples are also prone to heating caused by high radio frequency (RF) power pulses, especially during long proton decoupling. KcsA is stable at room temperature for extended periods of time in nonionic detergents, such as DM6 and maintains the tetrameric form even at relatively high temperatures (up to approximately 65 oC). For most membrane proteins the overexpression and purification is still a challenge. KcsA can be expressed in E. coli and purification details have been worked out 1. Methods Sample Preparation Uniformly 13C, 15N enriched KcsA (~8.8 mg/ml) was overexpressed and purified as previously described by (REF) mutations (Maybe brief description of purification??) ………..…………………………………………………………………………………… 4 …….. U-13C, 15N KcsA from S. lividans was expressed in E. coli (strain??) in 4 mg yield per liter of labeled minimal medium. After purification, protein purity was checked by Tricine-sodium dodecyl sulfate polyacrylamide gel electrophoresis (Tricine-SDSPAGE) according to the method of by Schagger and von Jagow 36. The protein bands were visualized on a 10-20% gradient gel by silver staining. The protein sample was stored at 4 ºC in 50 mM potassium phosphate buffer (pH 6.0), 50 mM KCl, 0.01 mM sodium azide, 10 mM ethylenediaminetetraacetic acid (EDTA), and 10 mM n-Dodecyl D-maltoside (DM). Protein concentration was determined by UV measurements at 280 nm ( 34,850 M-1 cm-1). Approximately 2.3 mg of the protein was precipitated at room temperature by mixing equal volumes of protein solution and precipitant (50% PEG 1000, 50 mM Tris, 100 mM KCl, pH 7.4). After precipitation the protein was stored at 4 oC. Ethylene glycol and KCl were added to the solution to a final concentration of 15% (w/v) and 100 mM, respectively. The precipitate was then collected by centrifugation at 25,000 g (16,400 rpm) and transferred to a 4 mm Bruker NMR rotor. 2D 13C-13C homonuclear correlation spectra (DARR 37) were acquired at 750 MHz (Bruker Avance DRX-750 spectrometer). After data collection, approximately 1.2 mg of this sample was repacked into a 4 mm Varian NMR rotor. The K+ concentration was adjusted to approximately 110 mM before repacking. 2D 13C-13C homonuclear correlation spectra (DARR) and 3D inter-residue (NCOCX) and intra-residue (NCACX) spectra of the repacked sample were acquired on a Varian Infinity Plus 600 spectrometer. Solid State NMR: Pulse Sequences and Acquisition Parameters The techniques and pulse sequences used are similar to those successfully applied for the assignment of backbone and sidechains of ubiquitin 20, 38. The assignments of KcsA are based on four spectra: two 2D 13C-13C homonuclear DARR spectrum acquired at 750 MHz and 600 MHz and two 3D DCP-DARR spectra recorded at 600 MHz. A cartoon diagram of the pulse sequences is shown in the Supplementary Materials. 2D 13C-13C homonuclear correlation spectra of KcsA were acquired by the DARR (Dipolar Assisted Rotational Resonance) 37 method. After the initial ramped magnetization transfer 39 from 1H to 13C by cross polarization at the Hartmann-Hahn condition, simple proton assisted spin-diffusion mediates homonuclear polarization transfer. During the mixing time the proton field strength is matched with the rotational frequency of the rotor. 3D NCACX and NCOCX spectra were acquired by the DCPDARR sequence 20, which combines a selective double cross polarization (DCP) 40, 41 sequence and DARR. The double cross-polarization sequence (DCP) was used to selectively direct the polarization from 15N to 13CA or 13CO, which was then transferred to other 13C nuclei by DARR. Acquisition Parameters 2D 13C-13C homonuclear correlation spectra of KcsA were acquired by the DARR 37 method on a Bruker Avance DRX-750 spectrometer. The spectrometer was operating at Larmor frequencies of 750.22 MHz for 1H and 188.660969 MHz for 13C. The experiments were carried out at 250 K and at 10 kHz spinning frequency. Note that 250 K was the temperature of the air cooling the sample; the actual sample temperature was higher due to frictional forces from magic angle spinning and RF pulses. The sample 5 heating is not well calibrated at this point; it is estimated to be approximately 20 K higher than the apparent temperature. H 67.6 kHz, C 45.5 kHz, 0.8 ms Cp conditions ……………………………………….. There were 2600 points collected in the direct and 1024 points in the indirect dimension. The FID’s were acquired for 18.2 ms in the direct dimension and 12.8 ms in the indirect dimension. The spectrum was collected for 28 hours (32 scans with 3 s pulse delay). During mixing the proton field strength was matched to the spinning frequency. The mixing time was 13 ms, which was sufficient to allow the observation of 2-3 bond transfers for most amino acids. 2D 13C-13C homonuclear correlation spectra were also collected on a Varian Infinity Plus 600 spectrometer at 253K and a 13 kHz spinning frequency. The DARR mixing time was increased to 200 ms in order to identify medium range contacts. Cross polarization was achieved by a 67 kHz square 1H pulse at 599.26325 MHz and 48- 58 kHz tangent ramped 13C pulse at 150.70005 MHz for 1 ms. There were 1024 points collected both in the direct and indirect 13C dimensions for 17 ms and 13 ms, respectively. During acquisition 71 kHz TPPM decoupling pulse was applied on the 1H channel. During the 200 ms mixing the proton field was matched to the spinning frequency. There were 192 scans collected with 3 s pulse delay. 3D 13C/15N chemical shift correlation solid-state NMR spectra were acquired on a Varian Infinity Plus 600 spectrometer with a spinning frequency of 13 kHz and an apparent sample temperature of 243 K. The spectrometer was operating at Larmor frequencies of 599.290000 MHz for 1H, 60.732297 MHz for 15N, and 150.717491 MHz and 150.698970 MHz for 13CO and 13CA, respectively. Two 3D experiments were carried out: an NCOCX experiment for inter-residue backbone-backbone and backbonesidechain correlations and an NCACX experiment for intra-residue correlations. For the 3D NCOCX inter-residue correlation experiment, 1H-15N crosspolarization was carried out using a tangent radio frequency (RF) pulse on the 15N channel, with RF field of 53.7 kHz for the 1H channel and 33.7 kHz (at the center of the ramp, with 10 kHz ramp) for the 15N channel. The contact time was 0.7 ms. The 13CO was selectively polarized by a 45.9 kHz 15N pulse and a 30.4 kHz 13C pulse with a slight ramp of 2.0 kHz for 2.0 ms. Homonuclear 13C-13C mixing was accomplished using the DARR pulse sequence with a mixing time of 19.23 ms. There were 1024 points collected in the direct 13C dimension, 48 points in the indirect 15N dimension, and 32 points in the indirect 13C dimension. Data were recorded for 16.89 ms in the direct dimension, 7.38 ms in the indirect 15N dimension, and 4.96 ms in the indirect 13C dimension. The spectrum was collected for 6.9 days, in 8 blocks with 20.8 hours duration for each block. For the NCACX intra-residue correlation experiment, 1H-15N cross-polarization was carried out using a tangent radio frequency field on the 15N channel, with the field strength of 53.7 kHz for 1H and 34.5 kHz for 15N (at the center of the ramp), with a 10 kHz ramp for a duration of 0.7 ms. A ramped radio frequency field was used on the 13C channel with the field strength of 28.8 kHz for the 15N and 17.1 kHz for the 13CA (at the center of the ramp), ramp size of 1.4 kHz, and a contact time of 2.0 ms. Homonuclear 13 13 C- C mixing was accomplished using DARR sequence with a mixing time of 19.20 ms. There were 1024 points collected in the direct 13C dimension, 48 points in the indirect 15N dimension, and 48 points in the indirect 13C dimension. The FIDs were 6 recorded for 16.89 ms in the direct dimension, 7.38 ms in the indirect 15N dimension, and 3.72 ms in the indirect 13C dimension. The spectrum was collected for 6.5 days, in 12 blocks with 13.0 hours duration for each block. NMR Data Processing All data were processed with NMRPipe 42; analysis and assignments of the 2D and 3D data sets were carried out using Sparky version 3.1 43. All spectra were processed at least two different ways: (i) by applying only sinebell apodization in each dimension; (ii) by applying exponential multiplication in order to enhance the signal-to-noise, which proved to be valuable for the assignment of weak isolated peaks. Both dimensions of the 2D DARR spectra were apodized by sinebell window function and zero filled to 4096 points. The spectra were also processed by applying exponential line broadening in both the direct and indirect dimensions (50 and 100 Hz respectively), then the FIDs were zero filled to 4096 points before Fourier transform. Both the NCACX and NCOCX 3D spectra were processed with sinebell apodization function in both the direct and indirect dimensions. Alternatively, the NCACX 3D spectrum was also processed with exponential line broadening: 100 Hz in the observed 13C dimension, 40 Hz and 100 Hz broadening in the indirect 15N and 13C dimensions respectively. For the NCOCX 3D spectrum, 100 Hz, 20 Hz, and 50 Hz exponential line broadening was applied in the observed 13C and indirect 15N and 13C dimensions respectively. In each case the direct carbon dimension was zero filled to 4096 points, while the indirect 13C and 15N dimensions were both zero-filled to 256 points. For each experiment, the carbon dimension was referenced externally to TMS using the 13C adamantane methylene peak at 38.56 ppm, which was then adjusted to DSS assuming a 1.7 ppm chemical shift difference 44. The nitrogen dimension was referenced to 15N labeled ammonium chloride………….. Results and Discussion There were two 2D 13C-13C DARR mixing spectra acquired: a DARR spectrum with 13 ms mixing time which allowed for the observation of two bond transfers for most amino acids and a spectrum with 200 ms mixing time for in order to observe medium range contacts. The short mixing time 2D 13C-13C DARR mixing spectrum is usually the first 2D spectrum recorded. These spectra are used to confirm residue types and the secondary structure elements of the protein (i.e. helical nature). Since 13C chemical shifts are sensitive to secondary structure changes, the 2D 13C-13C DARR mixing spectra were also used to confirm sample integrity throughout the course of NMR measurements. As reported by Zech et al.32, the majority of sequential contacts (<3.8 Å) can be observed at 200 ms. Due to the additional crosspeaks spectral crowding can be considerable, therefore long mixing times are usually employed for proteins with selective labeling (i.e. proteins purified from bacteria grown with 1,3-13C or 2-13C glycerol). For KcsA the 200 ms spectra were very congested for most regions as expected for a U-13C labeled 160 amino acid protein. However, some regions provided very useful insights into the assignments. As the initial step of the assignment, the amino acid type of most cross peak clusters is identified according to their typical chemical shift and connectivity. Since most of the protein has helical secondary structure, there are very congested areas. On 7 the other hand, there are some well resolved peaks in the spectrum either because there are only a few residues of a certain type of amino acid, or because amino acids have a secondary structure other than right handed alpha-helical. Because of the unique chemical shifts, these ‘outliers’ can be considered as a ‘finger-print’ region and were used to monitor sample integrity. One example for sparse amino acid type is isoleucine, since the KcsA sequence contains only three of them (I38, I60, I100). The C and the sidechain peaks for two isoleucines can be clearly assigned in the 13 ms DARR spectrum, however the CO peaks could not be assigned unambiguously. The third isoleucine was not observed under our experimental conditions. In the 200 ms DARR spectrum provided additional, well resolved CD-CO peaks which confirmed the CO assignments unambiguously, as illustrated in the Figure xx in the Supplementary Materials. According to chemical shift prediction by ShiftX45 based on the crystal structure of KcsA4 (PDB accession code: 1K4C), two of the three isoleucines should have chemical shifts which correspond to alpha-helical confirmations, and I60 should have a chemical shifts which corresponds to beta-sheet. Therefore, although not confirmed by sequential backbone walk, the isoleucine with beta-sheet like chemical shift is most likely I60. Other unique examples of outliers were found among the valines. Two valines outliers could be readily identified in the DARR spectra according to their connectivity. Since these valines have such different chemical shifts from the other 14 valines, it was a likely hypothesis that these valines are part of the pore region. We pursued this hypothesis and found plausible backbone walks in the 3D spectra. Based on the backbone walk, we assigned the valine outliers as V76 and V84. Figure 2 illustrates the two valine outliers in the DARR spectra, and Figure 4 shows the strip plots of the backbone walk form the 3D experiments. Site-specific assignments for the Y82, P83, and V84 carbonyls were also supported from the 200 ms DARR spectrum. The P83 CD correlation with P83 CO, and the CO of the neighboring residues Y82 and V84 were identified. The CO chemical shifts matched the assignments obtained from the 3D spectra (Figure 5). The backbone assignment strategy in the 3D spectra is very analogous to that used in solution NMR. The polarization transfer pathway implemented by the 3D DCPDARR experiments is presented in the Supplementary Materials. In the NCACX experiment, the polarization is selectively transferred from 15N to the 13CA of the same residue by DCP, which is then distributed to the 13CO and also to the side chains by the DARR sequence. Intra-residue correlations are obtained in this experiment. The NCOCX experiment is very similar, however the transfer is optimized for 15N to the 13CO of the preceding residue, which is then distributed to the 13CA and the sidechains by DARR. The NCOCX experiment provides inter-residue correlations thus making the ‘backbone walk’ feasible. In the 3D spectra the 15N dimension dramatically improved the resolution for some of the very congested regions of the 2D 13C-13C DARR spectrum. This is illustrated in Figure 3 for the alanine CA-CB region. The KcsA sequence has 24 alanines, and the CA-CB crosspeak region can be readily identified in the DARR spectrum (Figure 3A) based on their typical alpha-helical alanine chemical shifts (55.2 ppm for CA and 19.7 for CB 46). However, only one peak can be resolved in the DARR spectrum. In the NCOCX 3D spectrum the congestion is relieved by the combination of 8 15 N chemical shift dispersion of the proceeding residues and the alanine 13CO dispersion. Unique sidechain shifts can further improve resolution as illustrated in Figure 3B. Based on the 15N-13CA-13CO chemical shifts only three alanines can be resolved, however the CB shifts further improve the resolution and four peaks can be identified. Sidechains also played an important role in confirming assignments for the backbone walk. In Figure 4, strip plots of sequential assignments are displayed for some residues where not only CA-CO backbone cross peaks but also the sidechains support the identity of the specific amino acids. These side chains were also confirmed by the 2D 13 13 C- C DARR spectrum. Assignment efficiency was also limited by the particularly weak peaks in regions with 15N chemical shift lower than 115 ppm or higher than 130 ppm. Usually these are the regions where outliers are found which often facilitate the assignment process. For the identification of peaks in these regions (i.e. peaks with glycine or proline 15N correlations), spectra processed with exponential line broadening were used in almost each case. In summary, sequential peak assignments were determined for residues V76-D80 and P83-C90 of KcsA. During the assignment process, peaks identified in the 3D spectra were cross-checked against peaks observed in the 2D 13C-13C DARR spectrum. There was a close agreement between the 2D and the 3D spectra. The backbone walk is illustrated by the strip plots in Figure 4. The assigned regions are underlined in the KcsA sequence in Figure 1. Both of these regions are part of the pore, and V76-D80 is also part of the selectivity filter. The assigned regions are highlighted in the crystal structure of KcsA (PDB: 1BL8) in the Supplementary Materials. Although these assignments need to be confirmed by future experiments, there is a high probability that at least some of them are correct due to the lack of supportive evidence for alternate lists. Improving resolution in future experiments can facilitate assignments. Narrow linewidths are especially important for mostly alpha-helical membrane proteins where backbone carbons exhibit very congested spectra due to conformational similarity. 3D pulse sequences implemented with homonulear 13C J decoupling (i.e. DANTE sequence) during evolution in the indirect dimension could also significantly improve resolution, as has been demonstrated for ubiquitin 47. Selective isotope labeling can also reduce homonuclear dipolar couplings; the use of site directed biosynthetic labeling has been demonstrated before to be useful for ssNMR assignment studies of the -spectrin SH3 domain 34 and for the most recent studies of LH2 26, 48: E. coli grown in minimal medium supplemented with [2-13C] or [1,3-13C] glycerol produces proteins with parse labeling 49, 50 in a specific pattern that most labeled carbons have unlabeled neighbors. This study forms the basis for other biophysical studies which could explore the ion conduction. Solid state NMR studies do not require diffraction quality crystals, therefore the precipitation conditions are greatly flexible for the protein. Since KcsA can be precipitated under various conditions, structural changes induced by pH, salt concentration or toxin binding can be easily detected by observing perturbed chemical shifts. Conclusions 9 The assignment of integral membrane proteins by solid state NMR is still a difficult challenge. Our study forms the basis for a broader investigation for the structure and mechanism of ion conduction of the KcsA potassium ion channel, an integral membrane protein by solid state NMR. The prerequisite for NMR based structure determination is the assignment of the observable 13C and 15N resonances to corresponding nuclei. As the first step of the study, we established solid state NMR sample preparation method for a homogenous sample. Solid state NMR sample preparation method is crucial in obtaining narrow line widths thus resolving peaks, as has been underlined by several studies (REF SH3 and ubiquitin)27, 28. The feasibility of the sequential, site-specific assignments of KcsA backbone and sidechains is well demonstrated in this study. The hypothetical assignments of the pore region and selectivity filter were based on a combination of 2D and 3D NMR spectra acquired at high field. This study demonstrates that solid-state NMR methods provide a promising biophysical approach for many important and challenging biological systems. 10 11 Cited References 1. Doyle, D. A.; Cabral, J. M.; Pfuetzner, R. A.; Kuo, A. L.; Gulbis, J. M.; Cohen, S. L.; Chait, B. T.; MacKinnon, R., The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, (5360), 69-77. 2. Long, S. B.; Campbell, E. B.; MacKinnon, R., Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, (5736), 897-903. 3. Jiang, Y. X.; Lee, A.; Chen, J. Y.; Cadene, M.; Chait, B. T.; MacKinnon, R., The open pore conformation of potassium channels. Nature 2002, 417, (6888), 523-526. 4. Zhou, Y. F.; Morais-Cabral, J. H.; Kaufman, A.; MacKinnon, R., Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 angstrom resolution. Nature 2001, 414, (6859), 43-48. 5. Martin, R. W.; Zilm, K. W., Preparation of protein nanocrystals and their characterization by solid state NMR. Journal of Magnetic Resonance 2003, 165, (1), 162174. 6. Cortes, D. M.; Perozo, E., Structural dynamics of the Streptomyces lividans K+ channel (SKC1): Oligomeric stoichiometry and stability. Biochemistry 1997, 36, (33), 10343-10352. 7. Hong, M., Solid-state NMR determination of C-13 alpha chemical shift anisotropies for the identification of protein secondary structure. Journal of the American Chemical Society 2000, 122, (15), 3762-3770. 8. Huster, D.; Yamaguchi, S.; Hong, M., Efficient beta-sheet identification in proteins by solid-state NMR spectroscopy. Journal of the American Chemical Society 2000, 122, (46), 11320-11327. 9. Luca, S.; Filippov, D. V.; van Boom, J. H.; Oschkinat, H.; de Groot, H. J. M.; Baldus, M., Secondary chemical shifts in immobilized peptides and proteins: A qualitative basis for structure refinement under Magic Angle Spinning. Journal of Biomolecular Nmr 2001, 20, (4), 325-331. 10. Heginbotham, L.; Abramson, T.; Mackinnon, R., A Functional Connection Between The Pores Of Distantly Related Ion Channels As Revealed By Mutant K+ Channels. Science 1992, 258, (5085), 1152-1155. 11. Heginbotham, L.; Lu, Z.; Abramson, T.; Mackinnon, R., Mutations In The K+ Channel Signature Sequence. Biophysical Journal 1994, 66, (4), 1061-1067. 12. http://www.rcsb.org/pdb/holdings.html. 13. http://pdbtm.enzim.hu. 14. McDermott, A.; Polenova, T.; Bockmann, A.; Zilm, K. W.; Paulsen, E. K.; Martin, R. W.; Montelione, G. T., Partial NMR assignments for uniformly (C-13, N-15)enriched BPTI in the solid state. Journal of Biomolecular NMR 2000, 16, (3), 209-219. 15. Pauli, J.; Baldus, M.; van Rossum, B.; de Groot, H.; Oschkinat, H., Backbone and side-chain C-13 and N-15 signal assignments of the alpha-spectrin SH3 domain by magic angle spinning solid-state NMR at 17.6 tesla. Chembiochem 2001, 2, (4), 272-281. 16. van Rossum, B. J.; Castellani, F.; Rehbein, K.; Pauli, J.; Oschkinat, H., Assignment of the nonexchanging protons of the alpha-spectrin SH3 domain by two- and three-dimensional H-1-C-13 solid-state magic-angle spinning NMR and comparison of solution and solid-state proton chemical shifts. ChemBioChem 2001, 2, (12), 906-914. 12 17. van Rossum, B. J.; Castellani, F.; Pauli, J.; Rehbein, K.; Hollander, J.; de Groot, H. J. M.; Oschkinat, H., Assignment of amide proton signals by combined evaluation of HN, NN and HNCA MAS-NMR correlation spectra. Journal of Biomolecular NMR 2003, 25, (3), 217-223. 18. Hong, M., Resonance assignment of C-13/N-15 labeled solid proteins by two- and three-dimensional magic-angle-spinning NMR. Journal of Biomolecular NMR 1999, 15, (1), 1-14. 19. Igumenova, T.; McDermott, A. E.; Zilm, K. W.; Martin, R. W.; Paulson, E. K.; Wand, J. A., Assignments of Carbon NMR Resonances for Microcrystalline Ubiquitin. Journal of American Chemical Society 2004, (accepted for publication). 20. Igumenova, T.; Wand, J. A.; McDermott, A., Assignment of the Backbone Resonances for Microcrystalline Ubiquitin. Journal of American Chemical Society 2004, 126, (16), 5323-5331. 21. Bockmann, A.; Lange, A.; Galinier, A.; Luca, S.; Giraud, N.; Juy, M.; Heise, H.; Montserret, R.; Penin, F.; Baldus, M., Solid state NMR sequential resonance assignments and conformational analysis of the 2 x 10.4 kDa dimeric form of the Bacillus subtilis protein Crh. Journal of Biomolecular Nmr 2003, 27, (4), 323-339. 22. Fujiwara, T.; Todokoro, Y.; Yanagishita, H.; Tawarayama, M.; Kohno, T.; Wakamatsu, K.; Akutsu, H., Signal assignments and chemical-shift structural analysis of uniformly C-13, N-15-labeled peptide, mastoparan-X, by multidimensional solid-state NMR under magic-angle spinning. Journal of Biomolecular NMR 2004, 28, (4), 311-325. 23. Marulanda, D.; Tasayco, M. L.; McDermott, A.; Cataldi, M.; Arriaran, V.; Polenova, T., Magic angle spinning solid-state NMR spectroscopy for structural studies of protein interfaces. Resonance assignments of differentially enriched Escherichia coli thioredoxin reassembled by fragment complementation. Journal of the American Chemical Society 2004, 126, (50), 16608-16620. 24. Franks, W. T.; Zhou, D. H.; Wylie, B. J.; Money, B. G.; Graesser, D. T.; Frericks, H. L.; Sahota, G.; Rienstra, C. M., Magic-angle spinning solid-state NMR spectroscopy of the beta 1 immunoglobulin binding domain of protein G (GB1): N-15 and C-13 chemical shift assignments and conformational analysis. Journal of the American Chemical Society 2005, 127, (35), 12291-12305. 25. Egorova-Zachernyuk, T. A.; Hollander, J.; Fraser, N.; Gast, P.; Hoff, A. J.; Cogdell, R.; de Groot, H. J. M.; Baldus, M., Heteronuclear 2D-correlations in a uniformly C-13, N-15 labeled membrane-protein complex at ultra-high magnetic fields. Journal of Biomolecular NMR 2001, 19, (3), 243-253. 26. van Gammeren, A. J.; Hulsbergen, F. B.; Hollander, J. G.; de Groot, H. J. M., Residual backbone and side-chain 13C and 15N resonance assignments of the intrinsic transmembrane light-harvesting 2 protein complex by solid-state Magic Angle Spinning NMR spectroscopy. Journal of Biomolecular NMR 2005, 31, (4), 279. 27. Hiller, M.; Krabben, L.; Vinothumar, K. R.; Castellani, F.; van Rossum, B. J.; Kuhlbrandt, W.; Oschkinat, H., Solid-State Magic-Angle Spinning NMR of Outer Membrane Protein G from Escherichia coli. ChemBioChem 2005, 6, (9), 1679-1684. 28. Lorch, M.; Fahem, S.; Kaiser, C.; Weber, I.; Mason, A. J.; Bowie, J. U.; Glaubitz, C., How to Prepare Membrane Proteins for Solid-State NMR: A Case Study on the Alpha-Helical Integral Membrane Protein Diaglycerol Kinase from E. coli. ChemBioChem 2005, 9, (6), 1693-1700. 13 29. Huang, L. Solid state NMR spectral properties and partial sequential assignment of a membrane protein, the Light Harvesting Complex 1 (LH1). Columbia University, New York, 2005. 30. Rienstra, C. M.; Tucker-Kellogg, L.; Jaroniec, C. P.; Hohwy, M.; Reif, B.; McMahon, M. T.; Tidor, B.; Lozano-Perez, T.; Griffin, R. G., De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proceedings Of The National Academy Of Sciences Of The United States Of America 2002, 99, (16), 10260-10265. 31. Seidel, K.; Etzkorn, M.; Sonnenberg, L.; Griesinger, C.; Sebald, A.; Baldus, M., Studying molecular 3D structure and dynamics by high-resolution solid-state NMR: Application to l-tyrosine-ethylester. Journal Of Physical Chemistry A 2005, 109, (11), 2436-2442. 32. Zech, S. G.; Wand, A. J.; McDermott, A. E., Protein structure determination by high-resolution solid-state NMR spectroscopy: Application to microcrystalline ubiquitin. Journal of the American Chemical Society 2005, 127, (24), 8618-8626. 33. Seidel, K.; Etzkorn, M.; Heise, H.; Becker, S.; Baldus, M., High-Resolution Solid-State NMR Studies on Uniformly [13C, 15N]-Labeled Ubiquitin. ChemBioChem 2005, 9, (6), 1638-1647 . 34. Castellani, F.; van Rossum, B.; Diehl, A.; Schubert, M.; Rehbein, K.; Oschkinat, H., Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 2002, 420, (6911), 98-102. 35. Lange, A.; Becker, S.; Seidel, K.; Giller, K.; Pongs, O.; Baldus, M., A concept for rapid protein-structure determination by solid-state NMR spectroscopy. Angewandte Chemie-International Edition 2005, 44, (14), 2089-2092. 36. Schagger, H.; von Jagow, G., Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Analytical Biochemistry 1987, 166, (2), 368. 37. Takegoshi, K.; Nakamura, S.; Terao, T., C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chemical Physics Letters 2001, 344, (5-6), 631-637. 38. Igumenova, T. I.; McDermott, A. E.; Zilm, K. W.; Martin, R. W.; Paulson, E. K.; Wand, A. J., Assignments of carbon NMR resonances for microcrystalline ubiquitin. Journal of the American Chemical Society 2004, 126, (21), 6720-6727. 39. Metz, G.; Wu, X. L.; Smith, S. O., Ramped-Amplitude Cross-Polarization In Magic-Angle-Spinning Nmr. Journal Of Magnetic Resonance Series A 1994, 110, (2), 219-227. 40. Schaefer, J.; McKay, R. A.; Stejskal, E. O., Double-cross-polarization NMR of solids. Journal of Magnetic Resonance (1969) 1979, 34, (2), 443. 41. Baldus, M.; Petkova, A. T.; Herzfeld, J.; Griffin, R. G., Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Molecular Physics 1998, 95, (6), 1197-1207. 42. Delaglio, F.; Grzesiek, S.; Vuister, G. W.; Zhu, G.; Pfeifer, J.; Bax, A., NMRpipe - A Multidimensional Spectral Processing System Based On Unix Pipes. Journal Of Biomolecular Nmr 1995, 6, (3), 277-293. 43. Goddard, D. t.; Kneller, D. G. Sparky 3. San Francisco. 14 44. Wishart, D. S.; Bigam, C. G.; Yao, J.; Abildgaard, F.; Dyson, H. J.; Oldfield, E.; Markley, J. L.; Sykes, B. D., H-1, C-13 And N-15 Chemical-Shift Referencing In Biomolecular NMR. Journal Of Biomolecular Nmr 1995, 6, (2), 135-140. 45. Neal, S.; Nip, A. M.; Zhang, H. Y.; Wishart, D. S., Rapid and accurate calculation of protein H-1, C-13 and N-15 chemical shifts. Journal Of Biomolecular Nmr 2003, 26, (3), 215-240. 46. http://www.bmrb.wisc.edu/referenc/published/Ikura_cs_study/part1.html. In. 47. Igumenova, T. I.; McDermott, A. E., Homo-nuclear 13C J-decoupling in uniformly 13C-enriched solid proteins. Journal of Magnetic Resonance 2005, 175, (1), 11. 48. van Gammeren, A. J.; Hulsbergen, F. B.; Hollander, J. G.; de Groot, H. J. M., Biosynthetic site-specific 13 C labeling of the light-harvesting 2 protein complex: A model for solid state NMR structure determination of transmembrane proteins. Journal of Biomolecular NMR 2004, 30, (3), 267. 49. Hong, M.; Jakes, K., Selective and extensive C-13 labeling of a membrane protein for solid-state NMR investigations. Journal of Biomolecular Nmr 1999, 14, (1), 71-74. 50. LeMaster, D. M.; Kushlan, D. M., Dynamical mapping of E-coli thioredoxin via C-13 NMR relaxation analysis. Journal of the American Chemical Society 1996, 118, (39), 9255-9264.