Lecture 2, Soil Solution - Soil and Environmental Biogeochemistry
advertisement

1
GES 166/266, Soil Chemistry
Winter 2000
Lecture Supplement (2)
Chemistry of the Soil Solution
II
SOIL SOLUTION
II-1, The concept of Activity
An element or a molecule that has a formal charge is termed an ion, e.g., Ca2+.
Positively charged ions are termed cations and negatively charged ions are called anions.
Inorganic ions are the predominant components of soil solution that we will discuss.
When considering a species in solution most of us think in terms of
concentrations, which is defined as a quantity of a the species for a given volume of the
solution; an example would be mg/L or mols/L. While concentrations are informative,
they do not provide the needed information we need in order to assess the 'reactivity' of an
ion in solution. Nevertheless, they are widely reported because they are easily
measurable. So, we should ask ourselves two questions: (1) if concentrations are not
good for determining the reactivity of the solution then what unit is? and (2) how do we
obtain these units if we generally measure solutions for their species concentrations?
The answer to question (1) is that we define a unit of measurement termed
ACTIVITY. Activity, rather than concentration, is a unit that defines the reactivity of an
ion within the solution. I can most easily give the reasoning for this by analogy. Suppose
that you have a room which is 10 m x 10 m x 10 m, and 5 seven year old female children
are in the room. Your concentration of female children is 5 / room (or 5 / 103 m3).
Now, let's assume that each child has the same energy and that they are the only ones in
the room; then if we define their activity as their movement around the room, their
activity would equal their concentration at a value of 5 / room. Now let's fill the room
with 25 adults, including their parents, and measure their activity. The kids would no
longer be free to move around the room unimpeded; they would be restricted by the
physical presence of the adults and while their concentration is still 5 / room their activity
would be considerable less. In fact, we could fill the room with 25 seven year old female
children and their activity would not necessary equal their activity if they impede the
movement of one and other.
Let's move back to a real solution now. Ions that are in solution move around just
like children in a room. If their movement is not restricted then we could measure their
concentration and know their activity. When would this occur? Only in very, very dilute
solutions!!!! Under all other condition their concentrations and activities will NOT be
equal. We must, therefore, consider the 'make-up' of the solution in order to determine
their activity and hence reactivity.
With a general comprehension of activities and concentrations we need to now
introduce a few more formal terms:
2
Ideal State: In the thermodynamic consideration of solutions derivations are
based on an Ideal State. So what are ideal conditions. They are hypothetical situations in
which concentrations equal activities, so that you could measure the concentrations of a
solution and use these values directly to assess its reactivity. In soils, or any 'real' system,
ideal conditions do not exist. Soil solutions would be considered Non-Ideal since
concentrations and activities are not necessary equal.
Reference State: This is a conditions that can be achieved experimentally where a
system behaves as close to ideality as possible. What would this situation be for
solutions? (I just gave you the answer a few paragraphs ago) It is an infinitely dilute
solution; if their are very few ions in solution then their activities will just about equal
their concentrations (think of the kids in the room analogy).
Concentration: The mass of a species per unit volume of the solution.
Activity: The reactivity of a species per unit volume of solution.
The proceeding discussion gives us insight into our first question on the reactivity
of a system. But our second question, how do we determine activities, is not yet apparent.
Remembering that often we can only measure concentrations in solution then we must
use an equation in order to determine the activity of ions in solution.
Activity = a = M
[2.1]
where M = concentration (mols / L) and = activity coefficient
This equation states that the activity of a species is proportional to its
concentration, and that through a constant called an activity coefficient () we can
calculate activities directly from measured concentrations. But we need to know in
order to do this. Right now you probably are hoping I will provide a table and you can
just look up values of . I wish that were the case but unfortunately we need to introduce
a few more equations.
There is no exact equations for , only empirically based ones. These equations
have evolved to the point were they are accurate over a fairly broad range of conditions.
All equations that calculate activity coefficients are based on a solution parameter termed
Ionic Strength (I). The ionic strength is a measure of the total electric field strength that
an ion 'feels' in the solution. Think of it as a drag coefficient; if we are speaking of the
kids in the room analogy it would be the friction each child feels from everyone else
crowded into the room. Since ions have charges they will 'pull' on each other in solution
and thus restrict their motion and hence reactivity. The ionic strength is easily
determined by the follow equation:
Ionic Strength = I = 1/2 Mi Zi2
[2.2]
3
where M is again the concentration of an ion in solution and Z is its valence. From this
equation you should realize that I is summed over all of the ion constituents in solution.
Using this equation you would multiply the concentration (in mols / L) of a species by the
square of its valence, then do the same for a second ion and add it to the first; keep doing
this until all of the ion types have been considered and then take 1/2 of this value.
With the ionic strength defined we can now introduce various equations for the
activity coefficient:
Davies Equation:
log = – 0.511 Z2 {
I1/2 – 0.3 I}
1 + I 1/2
for I < 0.5
[2.3]
for I < 0.01
[2.4]
for I < 0.1
[2.5]
Debye-Hueckel Limiting Equation:
log = - 0.511 Z2 I 1/2
Extended Debye-Hueckel Equation:
0.511 Z 2 I1/2
log = – {
}
1 + B a I1/2
B is a temperature dependent constant (0.33 @ 25 °C)
a is an effective ion size parameter
For ionic strengths greater than 0.5 we need to use the Pitzer Equation; since most
soils do not have I values this large we will omit this more rigorous equation from our
discussion. The Davies equation is by far the most widely used equation for calculating
activity coefficients in soils.
Let's summarize the process we have described so far for determining activities.
First the solution is measured for the concentration of different species; then the ionic
strength is calculated; activity coefficients are calculated using an equations such as the
Davies equation. Finally, the activity is calculated. Unfortunately, there is one very
important factor that we have not yet introduced. In the following section, Ion-Ion
Interactions, we will consider this last factor and then provide the complete formula for
calculating activities. So, if I give you a homework or test question on calculating
activities you can't stop here, you need to read on and finish the next section!
There are a few exceptions where we can measure activities directly without
having to go through these calculations. The most notable case is pH measurements.
Here, the activity of the hydrogen ion, H+, is measured by an electrode that 'senses' the
reactivity or activity of this ion in solution. In fact, any ion specific electrode measures
activity rather than concentration. However, the most routine analytical methods such as
atomic absorption or inductively coupled plasma spectrophotometry measure
concentrations not activities.
II-2,
Ion-Ion Interactions
4
We have already introduced the concept that ions interact with each other by
defining the ionic strength of a solution. Now we need to consider more direct
interactions of ions with each other. In solution, we can broadly classify ions and their
interaction products into 3 groups:
Free Ions: These are ions that only have water coordinating them and would
exhibit their full charge. They are written as Ca2+, Na+, Cl-, NO3-, etc.... As I just stated,
these ions actually have water around them. Because of this you may see an ion such as
Fe2+ written as Fe2+•6H2O (termed hexaquo ferrous iron).
The number of waters
coordinating an ion is dependent upon a size factor that we will discuss in detail in the
next section of the class. For now realize that these ions do have water around them.
H2 O
H2 O
Fe
H2 O
H2 O
H2 O
H2 O
H2 O
Fe 2+
H2 O
H2 O
H2 O
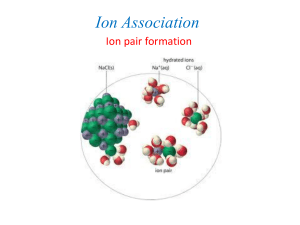
Ion Pairs: Ion pairs are fairly weak associations that form strictly from the
electrostatic attraction of ions with opposite charge. The water molecules surrounding an
ion that we just spoke of are not removed from the ions when they form ion pairs.
Because the water molecules are left coordinating the individual ions in the ion pair, this
association is called an 'outer-sphere' complex. The term outer-sphere means that the
association is outside the water sphere coordinating the ion. This is an important concept
that will keep reoccurring throughout the course, especially in the final section on the
solid/water interface. An example is Ca2+ combining with sulfate, SO42-, to form the
CaSO4o (aq) ion pair. Notice that the charge of two divalent ions is neutralized. What
effect do you think such species would have on the ionic strength of the solution?
5
H2 O
H2 O
Ca 2+
H2 O
H2 O
H2 O
H2 O
SO 4 2H2 O
H2 O
Ion Complexes: When ions are attracted to each other chemically or
electrostatically, and disrupt their individual hydration spheres (coordinating waters), an
inner-sphere complex is formed. We term these 'ion complexes'. They are a much
stronger association than occurs with ion pairs.
H2 O
H2 O
H2 O
Na +
FH2 O
H2 O
H2 O
Both the ion pair and ion complex are ion-ion interactions, but the ion pair is a
much weaker interaction. We can quantify the level of interaction, or the interaction
strength, with a thermodynamic parameter termed the Stability Constant (or Equilibrium
Constant), K. Depending upon how we write the reaction, the stability constant can be
expressed as an association or a dissociation constant; they are not equal in value so you
need to be aware of the direction in which the reaction is written. Typically, equilibrium
constants for ion complexes and ion pairs, as well as the solubility of a solid, are written
in terms of a dissociation reactions. As an example let's use the calcium sulfate ion-pair
again.
6
CaSO4o <=> Ca2+ + SO42-
[2.6]
KCaSO4o = (aCa2+) (aSO42-) / (aCaSO4o) = 10-2.274
[2.7]
Throughout this paper and in my lectures I will denote activities by parentheses () and
concentrations by brackets []. Please remember this since it is very important to
distinguish the difference between these to units.
A side note for your information, FYI: The equilibrium constant is related to
the energy of the reaction. This relationship is expressed most commonly in
terms of the Gibb's Free Energy (∆Grxn) of the reaction:
∆Grxn = - RT ln K
where R is the universal gas constant and equals 8.314 J / mol•K (or 1.987 cal
/ K•mol) and T is temperature in degrees Kelvin.
Energy
.
²G A
Free Io n
Activities
²G rxn
²G A = activation energy
²G rxn = Gibb's free energy
Ion Complex
Reaction Coordinate
II-3, Calculating Activities
After having addressed the formation of both ion pairs and complexes, we now
need to reconsider how we would calculate ion activities from measured total
concentrations. If there were no ion pairs or complexes then we could just take the
measured values of M (concentration) and determine I (ionic strength), then (activity
coefficient), and finally a (activity). If we do consider ion pairs and complexes then we
7
would simply use the activities just calculated to find the quantity of an ion pair or
complex from an equation such as that in [2.7]. But here lies a dilemma: if some of the
ions in solution reside as an ion pair then they would not influence the ionic strength, yet
in our original calculation of the activities we considered all the ions to be as free ions
and contribute to I. We have circular needs: we need to determine the activities after
considering the ion pairs and complexes but we need the activities in order to calculate
the ion pairs and complexes. So the big question is how do we do this? We will proceed
using an ITERATIVE process in which with each successive loop of the procedure we get
closer to the true answer and final converge to single values.
You first proceed without considering ion pairs or complexes, just as we have
already outlined, to get a first estimate of the activities. Use these activities to determine
a level of IPs or ICs. Next, you convert the IP or IC activities back into concentrations
and subtract these quantities from the total concentrations.
converting aIP to MIP: aIP / IP = MIP
[2.8]
(a rearrangement of Eqn [2.1])
note: for an uncharged species = 1, and so a = M; values
obtained from the Davies equation can be used for the specific
charged species (i.e., 1 or 2).
Use a simple mass balancing expression to subtract:
Mtotal = MFI + MIP + MIC
[2.9a]
MFI = Mtotal - MIP - MIC
[2.9b]
or
With the new values of the free ion (FI) recalculate I, , and a values. Then obtain
another estimate of the ion pairs and complexes, subtract them from the total elemental
quantities again and repeat this process. Continue this routine until the values do NOT
change significantly between successive loops.
The interactive process used to calculate activities is summarized below.
THE ITERATIVE PROCESS
i. Measure the total concentrations
1. Calculate I
2. Calculate
3. Calculate a
4. Use the a values in 3 to calculate IPs and ICs
5. Translate IP activities into concentrations
8
6. Subtract the IP and IC concentrations from the total elemental
concentrations
7. Go back to 1 using these new values to calculate I
*Continue this 'loop' until successive values of
significantly.
a
and IPs-ICs do not change
II-4, Hydrolysis Reactions
Water is a polar molecule in which partial positive charges are associated with the
and partial negative charges with the O2-. Of course we all remember that water
undergoes dissociation to some extent forming H+ and OH- as defined by its dissociation
constant (K = 1 x 10-14). Both cations and anions with a high charge to size ratio can
enhance this dissociation by cleaving a water; anions would combine with H+ and cations
with OH-. Such reactions are termed hydrolysis reactions because the water molecule is
'split' and lead to the formation of 'hydrolysis products'. Ions that tend to induce
hydrolysis are known as 'hydrolyzable ions'. Examples of hydrolyzable ions common to
waters and soil solutions are: Fe(III), Al, Cr(III), Co, and Mn.
Iron(III) and Al are two of the most prevalent ions in soil solutions that
extensively undergo hydrolysis reactions. Let's use Fe(III) as an example to demonstrate
these reactions. Because Fe(III) is a cation it will combine with OH-, so at low pH values
(say pH < 2), where there is a very low concentration of hydroxyl ions, we would expect
Fe(III) to exist as Fe3+•6H2O. However, as the pH increases Fe(III) with combine with
OH- to form its first hydrolysis product, Fe(OH)2+•5H2O.
H+s
H2 O
H2 O
H2 O
H2 O
Fe
H2 O
H2 O
H2 O
H2 O
Fe3+•6H2O
The first hydrolysis reaction of Fe(III) can be expressed:
H2 O
Fe OH
H2 O
H2 O
Fe(OH)2+•5H2O
9
Fe3+ + H2O <--> Fe(OH)2+ + H+
K = 10-3.05
[2.10a]
K = 1010.95
[2.10b]
or similarly we could write,
Fe3+ + OH- <--> Fe(OH)2+
An important point to take note of in either Reaction [2.10a or b] is that acidity is
produced (production of H+ or consumption of OH-). This means that if Fe3+ was added
to a water that was at pH 7 (neutrality), Fe would under go hydrolysis and thereby acidify
the water!!!! Why is this so important for soils? Suppose that Fe3+ or Al3+ were retained
on the surface of a clay particle in a soil, then the conditions changed and either ion was
displaced into the soil solutions. Upon entering the soil solution Al or Fe(III) would
immediately undergo hydrolysis and lower the pH: They would acidify the soil solution.
Before we move on any further I think it would be appropriate to review a few
rules on reaction expressions and conversion between different forms such as
those in Reaction [2.10]. Hydrolysis reactions can very easily be converted
between different forms of expression by coupling them with the water
dissociation reaction. Here is an example of the conversion between Reactions
[2.10a] and [2.10b].
Fe3+ + H2O <--> Fe(OH)2+ + H+
H+ + OH- <--> H2O
Fe3+ + OH- <--> Fe(OH)2+
log K
-3.05
14.0
10.95
Some things to notice are that: (i) when reactions are added the K values are
multiplicative, which means that in logarithm form they are additive, (ii) similar
terms on opposite sides of the reaction cancel, (iii) reactions can be reversed,
and (iv) when a reaction is reversed the K value is inverted (or in logarithm
form it simply changes sign).
Reaction [2.10] illustrates the first hydrolysis reaction for Fe(III); there are several more
hydrolysis reactions of Fe(III) that must be considered. The complete list of Fe(III)
hydrolysis reactions are listed as follows.
Fe3+ + H2O <--> Fe(OH)2+ + H+
K = 10-2.19
[2.11]
Fe(OH)2+ + H2O <--> Fe(OH)2+ + H+
K = 10-3.5
[2.12]
Fe(OH)2+ + H2O <--> Fe(OH)3o + H+
K = 10-7.4
[2.13]
Fe(OH)3o + H2O <--> Fe(OH)4- + H+
K = 10-8.5
[2.14]
10
Thus, Fe(III) can undergo 4 hydrolysis reactions, changing from a trivalent cation to a
monovalent anion. Ions with the ability to change the sign of their charge are called
amphoteric. Four H+ ions are generated for this complete hydrolysis sequence. While
all four of these reactions are possible, realize that it is the pH of the solution that will
dictate the extent of an ions hydrolysis. If the pH were already below 2, then Fe(III)
would not undergo any hydrolysis reactions. Aluminum and chromium(III) also undergo
the exact sample four reactions, with the only differences being the values of K for each
step.
You should also be aware that all cations can undergo such hydrolysis reactions,
which makes them acidic!!! Right! This brings up an interesting point; why are cations
such as Ca2+ referred to as 'base' cations. There is nothing basic about them: they
produce acidity not basicity (alkalinity)!
Ca2+ + H2O <--> Ca(OH)+ + H+
[2.15]
The term 'base cations' was established because generally cations such as Ca2+ are found
in higher pH soils while low pH soils are dominated by more highly hydrolyzable ions
such as Fe and Al. I will expand on this point in class to the point of exhaustion.
Anyone who has studied analytical or biochemistry should be familiar with pKa
values. Often, acid/base reactions are expressed in terms of pKa. This is advantageous
because when the pH of a solution equals the value of a reactions pKa, there will be equal
proportions of the product and reactant species. For example, considering again the first
hydrolysis reaction of Fe(III), Reaction [2.11:
Fe3+ + H2O <--> Fe(OH)2+ + H+
pK = 2.19
[2.16]
Therefore, at a pH value of 2.19 half of the Fe(III) should be present as Fe3+•6H2O and
the other half should be as Fe(OH)2+.
A final note is to reiterate that hydrolysis reaction occur not only for cations but
also for highly charged anions. A common anionic hydrolysis reaction that occurs in soils
is that of phosphate, PO43-.
-
log K
-2.15
[2.17]
H2PO4- <--> H+ + HPO42-
-7.20
[2.18]
-12.35
[2.19]
o
H3PO4 <-->
H+
+ H2PO4
HPO42- <--> H+ + PO43-
11
II-5, Acids and Bases
We just discussed that water can undergo dissociation to form H+ and OH-. In a
similar manner, complexes consisting of a cation-OH- or an anion-H+ can dissociate
making the former a base and the later an acid. We shall begin our discussion using acids
as an example, but you should realize that bases will have all of the same properties.
Acids and bases are classified as strong or weak, depending upon the degree of their
dissociation.
Strong acids would completely dissociate into an anion and H+. Examples would
include HCl, H2SO4, HF, HNO3, etc...
Weak acids will only partially dissociate in water, with the degree of dissociation
dependent upon their acidity constant. For example, consider a hypothetical monoprotic
acid, HA:
HA = H+ + A-
Ka
[2.20]
with
Ka = (H+) (A-) / (HA)
[2.21]
log Ka = log(H+) + log {(A-) / (HA)}
[2.22]
-log(H+) = - log Ka + log {(A-) / (HA)}
[2.23]
pH = pKa + log {(A-) / (HA)}
[2.24]
taking logarithms,
rearranging,
thus,
Based on equation [2.24] it should be very obvious why their are equal proportions of
acid and base with the solution pH = pKa. As another example, let's use a weak acid that
really exists, acetic acid.
CH3COOH <==> CHCOO- + H+
Ka = 10-4.8
pKa = 4.8
[2.25]
pH = 4.8 + log {(CHCOO-)/(CH3COOH)}
[2.26]
All this is just fine but you may be asking your self what do acids or bases have to do
with soil chemistry. Actually, nothing I just felt like discussing them because they have
12
been a hobby of mine since I was a small child. Just kidding. It turns out that weak acids
and bases are very important in soils. Molecules such as phosphate (PO43-), arsenate
(AsO43-), sulfate (SO42-) and many others are acids/bases and their speciation will be
dependent upon reactions such as those just illustrated. In addition, we shall see that the
surfaces of many minerals and particulate organic matter also act as weak acids/bases.
The examples of acids so far have considered only monoprotic acids. Actually, we have
already considered polyprotic acids; the deprotonation reactions for phosphoric acid are
portrayed in Reactions [2.17] - [2.19]. One last point I would like to address that may
already be obvious to all of you is the reversibility of these reactions. Consider the
phosphoric acid sequence described in Reactions [2.17-2.19], but this time suppose we
are starting under high pH conditions with a phosphate salt, e.g., Na3PO4. If the pH were
decreased the following reactions would apply:
PO43- + H2O <==> HPO42- + OH-
Kb1 = 1.4 x 10-2
[2.27]
HPO42- + H2O <==> H2PO4- + OH-
Kb2 = 1.59 x 10-7
[2.28]
H2PO4- + H2O <==> H3PO4- + OH-
Kb3 = 1.42 x 10-12
[2.29]
Therefore, PO43- is a base while H3PO4 would be an acid. I expect that most of you
knew this but it never hurts to review it quickly.
Buffering Capacities of Weak Acids and Bases
Buffering is defined as the resistance to change. Soils are extremely well buffered
systems with respect to many factors including pH or plant nutrients. Since we are
discussing acids/bases let's just stick with pH buffering for the moment. If soils were not
well buffered pH systems, then any small perturbation might severely limit or even
eliminate plant growth. Fortunately, the properties of ions such as phosphate or Fe(III)
will resist pH changes by undergoing association/dissociation reactions thereby
neutralizing added acidity of basicity. Weak acids/bases will have their strongest
buffering capacity near their pK values. Why? Simply because when the pH = pK half of
the complex exists as the base while the other half is in the acid form; this split allows a
large quantity of either species available to absorb H+ or OH-.
As I just stated in the proceeding section, mineral surfaces and organic matter
have weak acid/base properties. In fact, these materials exert a very strong influence on
the buffering of soils. Organic matter is particularly a very important pH buffer in soils.
Rather than having surface functional groups with a single pKa value, particulate organic
material will have groups with a spectrum of pKa values. This leads to a property called
"universal buffering", in which the OM will limit pH change over a very wide range of
pH values.
III
DISSOLUTION and PRECIPITATION
13
Solids can either be formed (precipitation) or destroyed (dissolution) depending
upon the conditions of the solution. Some of the most influential conditions affecting
dissolution/precipitation in soils are: ionic composition and concentration, pH, and
temperature. In addition, solution species that form strong complexes with the
constituents of a solid may enhance the dissolution of such solids. In this section we will
discuss the process of solid phase formation and destruction. Here we will emphasize the
precipitation/dissolution of solids from a solution stand point; in the next section of this
course we will take a detailed look at the solids themselves.
III-1, Mineral Dissolution
Some of the most common dissolution/precipitation reaction in soils are those of
carbonates, sulfates, and hydroxides. A few examples of such reactions are provided
below with their respective solubility constants.
log Ksp
CaSO4 (gypsum) <=> Ca2+ + SO42-
-4.60
[3.30]
CaCO3 (calcite) <=> Ca2+ + CO32-
-8.35
[3.31]
Fe(OH)3 (amorphous) <=> Fe3+ + 3OH-
-38.7
[3.32]
Al(OH)3 (gibbsite) <=> Al3+ + 3OH-
-33.0
[3.33]
-35.0
[3.34]
Fe(OH)2H2PO4 (strengite) <=> Fe3+ + 2OH- + H2PO4
Take a little time and look at each of these reactions. Which of the five solids listed
above is the least soluble? The iron hydroxide is the least soluble, followed by strengite
(an iron phosphate) and then gibbsite (an aluminum hydroxide). Gypsum would be the
most soluble of these solids. Just in case this isn't apparent to you, let's go through a
quick refresher discussion on the solubility of solid using gibbsite as an example. Based
on Reaction [3.33], the mass law expression would be:
Ksp = (Al3+) (OH-)3 / (Al(OH)3) = 10-33.0
[3.35]
For pure solids, we define their activities to equal unity (for our purposes you can assume
that all solids are in a pure state and so their activity will always equal 1). Therefore,
Ksp = (Al3+) (OH-)3 = 10-33.0
[3.36]
This equation, [3.36], tells us that if the solution is in equilibrium with gibbsite than the
activity of Al multiplied by the cube of the hydroxyl activity must equal 10-33.0. So, if
14
the solution had a pH of 6.0, then the pOH = 8.0 (pH and pOH must add to 14 by
definition, right?); if we put this into equation [3.36] we will determine the Al activity in
solution.
At pH 6.0:
Ksp = (Al3+) (10-8)3 = 10-33.0
(Al3+) = 10-33 / 10-24 = 10-9
[3.37a]
[3.37b]
You can see that there is very little aluminum in solution; assuming for the moment that
the activity coefficient for Al is 1 then the concentration of Al in solution would only be
1x10-9 M at pH 6. You should be able to see from this example that the smaller the Ksp
value the less soluble the solid.
III-2, Predicting Dissolution/Precipitation
2.1 Saturation Indices
In the proceeding discussion we looked at a solid in equilibrium with the solution.
What if we don't know whether equilibrium has been achieved? If it was not, then we
could not use equation [3.35] to determine the concentration of Al in solution. We need a
means for looking at a solution and determining if it is at equilibrium with a solid-phase,
is it expected the a the solid-phase will precipitate or dissolve.
Whether a mineral precipitates or dissolves depends upon the activity of its
constituents in solution and the energy of formation for the solid, which for our purposes
is represented by the Ksp value. The solution parameter representing the total activity of
the solid's constituents is defined by the Ion Activity Product.
Ion Activity Product (IAP): the 'total' activity of the ions that comprise a given
solid-phase.
For example, the IAP for gypsum would be:
CaSO4 (gypsum) <=> Ca2+ + SO42IAP = (Ca2+) (SO42-) / (CaSO4)
= (Ca2+) (SO42-)
[3.38]
[3.39]
You should now be asking yourself, "wait a minute, I thought that the Ksp for gypsum
was defined by equation [3.39]". Well, you are correct. The expression shown by
Equation [3.39] does represent that of the Ksp for gypsum. So what then is the difference
between the IAP and Ksp? Simple, the Ksp represents a condition were equilibrium is
achieved and the IAP is just the measured product of Ca2+ and SO42- activities in
solution at that time. Let me elaborate on this point a little further for clarification.
15
Suppose you take a beaker of water and dump a little gypsum in it; then after 2 months
have passed you come back and determine the activities of Ca2+ and SO42-. This, of
course, gives you the IAP of this solution with respect to gypsum. Now, if I also tell you
that within 1 month the gypsum will reach equilibrium with the solution, you should be
able to determine the activities of Ca2+ and SO42- strictly from the Ksp value and reaction
[3.38]. If I was correct and equilibrium was reached then the measured values of Ca2+
and SO42- should equal those deduced by the Ksp expression.
For a final consideration, suppose you measured Ca and SO42- only 1 week after
you added the gypsum to the water; at this time both ions should be at lower
concentrations than expected from the Ksp expression. From this we would also see that
the IAP is less than the Ksp for gypsum. This is a very important finding. It means that
we can use the IAP to determine (i) if we are at equilibrium, in which case the IAP = Ksp,
and (ii) whether we expect the solid to dissolve (IAP < Ksp) or precipitate (IAP > Ksp).
Why is this so? Let's turn back to an energy consideration. If the IAP represents the
'energy' of the ions in solution and the Ksp the energy of the solid we can make some
important deductions. If the energy of the solution is greater than that needed to form a
solid, then the solid will form: This is the case if the IAP exceeds the Ksp for a given
solid. Now, if the energy of the solution is less than that of the solid, the solid will
dissolve to increase the energy of the solution. This is when the IAP is less than the Ksp.
Finally, of the energy of the solution and solid are equal then there is not energy transfer
and we are at equilibrium (IAP = Ksp).
With this knowledge, we need to introduce a few terms commonly used to
represent the various condition just described. The energy balance of a solution with
respects to a specific solid is commonly given by a 'saturation index'.
Saturation Index (SI): a parameter that defines whether a solution is
oversaturated, undersaturated, or in equilibrium with a specific solid.
SI = log (IAP/Ksp)
[3.40]
for:
SI > 0
SI < 0
SI = 0
-->
-->
-->
oversaturation
undersaturation
equilibrium
Oversaturation: the condition in which the solution IAP for a solid exceed the
Ksp for that solid--precipitation of the solid is expected.
Undersaturation: the condition where the IAP for the solution is less than the
Ksp for a solid--dissolution of this solid is expected.
One last important point before we move on to an example. You should note that in the
definition of "Oversaturation" I said "precipitation of a solid is expected". I did not say
that it will occur, only that it is favorable energetically!!!! This is also true for whether a
16
solid will dissolve. The relationships described above for dissolution or precipitation
only consider the 'total' energy of the system. They do not say anything about the path the
system must go through to get to an endpoint. That is, they do not tell us anything about
how fact these dissolution or precipitation reactions will proceed. If they are very, very
slow then often we may not see any change in a system even if we determine that it is not
at equilibrium. Given enough time (which may mean a millennium) the system will reach
our predicted end point, but for some reactions this may not occur in our life times.
2.2 Stability Diagrams
A common method for assessing whether a solid-phase will form is through the
use of stability diagrams.
Stability Diagrams: a graphical plot of the activities of solution species that lead
to the favorable formation of solids based on thermodynamic (equilibrium)
considerations.
Because these diagrams assume equilibrium, you should realize that while they are useful
in predicting the possible formation of a solid they do not give proof that these solids will
form. We will come back to this point momentarily, but before we do let's look at how
we can construct such diagrams.
Consider the aluminum hydroxide mineral gibbsite, the solubility of which can be
expressed by the following reaction.
Al(OH)3 (gibbsite) <=> Al3+ + 3OH-
[3.41]
Rearranging this reaction so that the solubility is shown in terms of the proton we obtain,
Al3+
Al(OH)3 <=>
+
+
3H + 3OH = 3H2O
3OH-
Al(OH)3 (gibbsite) + 3H+ <=> Al3+ + 3H2O
log K
-33.8
3*(14)
8.2
[3.42]
Looking at reaction [3.42] you should already see that the solubility of gibbsite is
controlled by the activity of H+ (pH) and Al3+.
Ksp (gibbsite) = (Al3+) (H2O)3 / (Al(OH)3 gibb) (H+)3
108.2 = (Al3+) / (H+)3
[3.43]
[3.44]
17
changing to logarithms and rearranging,
8.2 = log (Al3+) - 3 log (H+)
[3.45]
log (Al3+) = 8.2 - 3pH
[3.46]
Using equation [3.46] we can construct a simple stability diagram by plotting log (Al3+)
vs pH. Such a plot will lead to a single line with a slope of -3 and an intercept of 8.2. If
we measure these two parameters, Al3+ and pH, in a solution and then plot this datum
point on our stability diagram we should be able to determine whether precipitation or
dissolution of gibbsite will result. If the point is above our gibbsite curve then we are
oversaturated and gibbsite should form, while dissolution should occur if the point falls
below this curve.
10
5
Precipitation
log (Al)
0
-5
-10
Dissolution
-15
-20
0
1
2
3
4
5
6
7
8
9
pH
Figure 3.1. Stability diagram for gibbsite calculated from Equation [3.46]. Based on this
thermodynamic prediction, if the coordinate of pH and log (Al) for a solution is above the
line then precipitation is expected; dissolution is expected if the point is below the line.
18
Gibbsite is a very simple example for a stability diagram. Kaolinite is a little
more complex and so we will consider this clay mineral next.
kaolinite
log K
+
3+
Al2Si2O5(OH)4 + 6H = 2Al 2H4SiO4 + H2O 5.71
[3.47]
in simplified form,
Ksp (kaolinite) = (Al3+)2 (H4SiO4)2 / (H+)6 = 105.71
[3.48]
switching again to log form,
2log (Al3+) + 2log (H4SiO4) - 6log (H+) = 5.71
[3.49]
3pH + log (Al3+) = 2.85 - log (H4SiO4)
[3.50]
or
In this case, we can use equation [3.50] to construct our stability diagram by plotting
{3pH + log (Al3+)} vs {log (H4SiO4)}.
log (Al) + 3pH
10
Gibbsite
9
8
Kaolinite
7
Solution
Species
6
5
4
-6
-5
-4
-3
-2
log H 4SiO 4o
Figure 3.2. Stability diagram for gibbsite and kaolinite with variables of pH, (Al), and
(Si). The area surrounding each mineral name represents conditions where each mineral
is the most stable product based on these 3 variables.
19
III-3, Complexing Agents and Chelation:
The dissolution of solids (or the inhibition of precipitation) can be enhanced by
both organic and inorganic complexing agents. By forming strong complexes with the
solid's cationic or anionic (ligand) constituents, dissolution of the solid can be promoted.
In equal proportions, organic complexing agents will exert a much stronger influence on
mineral dissolution than inorganic agents. Examples of organic complexing agents
include humic acids, fulvic acids, and many small molecular weight organic acids,
including citric, oxalic, salicylic, and many others. The predominant inorganic
complexing agents are the carbonate species: carbonate (CO32-), bicarbonate (HCO3-),
and carbonic acid (H2CO3). These species should always be considered when evaluating
the solubility of solids. In fact, plants exude such complexing agents in appreciable
amounts to promote mineral dissolution and thus enhance nutrient availability.
The process of complexing agent enhanced dissolution is also referred to as
'ligand promoted' dissolution. This is because most of the complexing agents are anions
that bind with the cationic constituents of solids; chemical and mineralogical terminology
is such that complex forming anions are termed ligands. If the ligand forms multiple
bonds, the resulting complex is termed a 'chelate'.
Chelation: a cation-ligand complex with multiple bonds between the ligand and
cation.
Chelated cations are particularly stable and unreactive due to the chemical and steric
properties of the complex. Again, the equilibrium constant should give you a good
indication as to the stability of the complex.
IV
COMPLEX EQUILIBRIA
Up to this point, most of the reactions we have considered only represent a small
and very simplified portion of the chemistry in a soil or natural surface water.
Nevertheless, this simple depiction is needed in order to learn the important basic
reactions that occur within these systems. In this section we will expand upon this
approach by considering multiple reactions and solving these quantitatively. We are
moving from a minute depiction of individual processes to a picture of the entire soil or
water system. I hope this is more gratifying, but of course with this applicability comes
the cost of a much more complex system that will be more rigorous to define.
IV-1, The Common Ion Effect
If an ion is introduced to a system from two different parent compounds (for
example, Ca being released by both calcite and gypsum), the amount of that ion can only
be determine by solving the both reactions that introduce it simultaneously. As an
20
example we will use a solution in which gypsum is first added followed by an ammonium
sulfate fertilizer. Our intent is to know the activities of Ca2+ and SO42- in solution so we
can determine their plant availability
First, we already know that the Ca entering solution from gypsum is defined by
the solubility expression of this solid.
CaSO4 (gypsum) <=> Ca2+ + SO42-
K = 10-4.6
[4.51]
Consider a condition in which the IAPgyp < Ksp, gyp
IAP ≈ 10-6 = (Ca2+) (SO42-)
so,
(Ca2+) = 1 x 10-3 M
(SO42-) = 1 x 10-3 M
We also know that some of the products, Ca2+ and SO42-, will combine to form the
CaSO4o ion pair.
Ca2+ + SO42- <=> CaSO4o
K =186
K = 186 = (CaSO4o) / (Ca2+) (SO42-)
[4.52]
[4.53]
(CaSO4o) = 1.86 x 10-4 M
An important point to consider is that even though some of the Ca2+ and SO42- will
combine to form the ion pair, the activities of the free ions will remain constant. Why?
Because as some of the free ions combine and form the ion pair, gypsum will undergo
further dissolution; if we are at equilibrium the activities of Ca2+ and SO42- will obtain
values that satisfy equation [4.51]. As long as there is excess gypsum in the system we
can always determine the activities of Ca2+ using equation [4.53]. We should now be
able to write a mass balance expression for the total Ca and SO4 in solution.
(Ca)T = (Ca2+) + (CaSO4o) = (1x10-3)+(1.86x10-4) = 1.186 x 10-3
[4.54]
(SO4)T = (SO42-) + (CaSO4o) = (1x10-3)+(1.86x10-4) = 1.186 x 10-3 [4.55]
Now we will complicate matters a little more by considering the addition of (NH4)2SO4,
with [SO42-] = 0.001 M. If you are wondering whether a situation like this could ever
really happen consider a small lake or pond in a semi-arid agricultural region. Often such
21
waters will have gypsum precipitated in their sediments so that the levels of Ca in
solution are dictated by the solubility expression of reaction [4.52]. Now, if a farmer
adds N to his/her fields as ammonium sulfate (which is commonly done), then we have
exactly the situation we are considering here.
Back to our example and the addition of ammonium sulfate. Since 0.001 M
sulfate was just added we have a new mass balance expression to replace equation [4.55].
gypsum amm-sulfate
(SO4)T = (SO42-) + (SO42-) + (CaSO4o)
= (1x10-3)+ (1x10-3) + (1.86x10-4) = 2.186 x 10-3
[4.56]
Rearranging equations [4.54] and [4.56] we can obtain expressions for each of the free
ions.
(Ca2+) = (1.186 x 10-3) - (CaSO4o)
[4.57]
(SO42-) = (2.186 x 10-3) - (CaSO4o)
[4.58]
And now for the good part, we take equations [4.57] and [4.58] and substitute them into
equation [4.53].
(CaSO4o) = 186{(1.186x10-3 - CaSO4o) (2.186x10-3 - CaSO4o)
[4.59]
= 4.82 x 10-4 - 0.627(CaSO4o) + 186(CaSO4o)2
[4.60]
0 = 4.82 x 10-4 - 1.627(CaSO4o) + 186(CaSO4o)2
[4.61]
Solving the quadratic equation in [4.61] gives the solution.
(CaSO4o) = 2.07 x 10-4 M
Substituting this value of the ion pair into equations [4.57] and [4.58] gives the complete
solution to our problem and fulfills our objective.
(Ca2+) = 8.79 x 10-4 M
(SO42-) = 18.9 x 10-4 M
IV-2, Carbonate Chemistry
Carbonate chemistry is extremely influential in soil, aqueous, and marine chemistry.
Even many small marine animals make use of carbonate chemistry to produce their
exoskeletons with the precipitation of aragonite. Arid soils will in particular be affected
22
by carbonate chemistry; even the weathering of primary minerals to form soils is
profoundly influenced by the carbonate system. The carbonate system is also one of the
predominant buffers within surficial waters and in biological systems--it is the key to
buffering within our blood!
IV-2.1, Components and Reactions of the Carbonate System
The carbonate system is particularly complex because it involves a gas phase
species (CO2, g), 4 solution species (CO2 (aq), H2CO3, HCO3-, and CO32-), and a solid
phase component (CaCO3). In practice, we will combine the CO2 (aq) and H2CO3 into a
single term H2CO3*, giving us 3 solution species to consider. A schematic representation
of the complete carbonate system is shown below.
In the atmosphere, CO2 comprises 0.03% (PCO2 = 3 x 10-4 atm) of the total
composition. In soils, occluded pores may limit gas exchange with the atmosphere and
microorganism activity can then lead to partial pressures greater than ten times the
amount found in the atmosphere. We will need to consider variable partial pressures of
CO2 (g) since soils can have a wide range of levels of this gas.
There are several important reactions that pertain to the carbonate system. The
primary equilibrium reactions are specified below.
H2 O
CO2 (g) <=> CO2(aq)
[4.62]
H2CO3 = CO2(aq) + H2O
[4.63]
H2CO3 = H+ + HCO3-
[4.64]
HCO3- = H+ + CO32-
[4.65]
CaCO3 (s) = Ca2+ + CO32-
[4.66]
23
These equilibrium reactions lead to the expressions that will allow us to define the
speciation of the carbonate system. We first look at the exchange of aqueous-phase with
gaseous-phase carbon dioxide.
CO2 (g) <=> CO2(aq)
[4.67]
CO2(aq) = KH PCO2
[4.68]
where,
KH = Henry's Law Constant = 0.033 M (at 25 °C)
[4.69]
Therefore, in an atmosphere completely of CO2 (g), [CO2 (aq)] = 0.033 M. Once
dissolved in water, CO2 (aq) can combine with the water molecule to form carbonic acid.
H2CO3 = CO2(aq) + H2O
K = 102.8
[4.70]
However, as exhibited by the value of the equilibrium constant, only a small percentage
of CO2(aq) actually combines with water to from H2CO3--less than 0.2%! Operationally,
it is difficult to distinguish between the two, so that in practice H2CO3 and CO2(aq) are
combined and treated as one: H2CO3*.
H2CO3* = H2CO3 + CO2(aq)
[4.71]
CO2 (g) <=> H2CO3*
[4.72]
KH = (H2CO3*) / PCO2
[4.73]
With the formation of H2CO3*, the next consideration is its dissociation.
H2CO3* = H+ + HCO3K1 = (H+)(HCO3-) / (H2CO3*)
K1 = 10-6.36
[4.74]
pK1 = 6.36
You should note that H2CO3* is a much weaker acid than true H2CO3, as shown by their
dissociation constants (or pKa). Since H2CO3* is a diprotic weak acid, we next look to
the second dissociation reaction; the deprotonation of bicarbonate (HCO3-) leading to the
formation of the carbonate ion (CO32-).
HCO3- = H+ + CO32K2 = (H+)(CO32-) / (HCO3-)
K2 = 10-10.33
pK2 = 10.33
[4.75]
24
Using equations 4.74 and 4.75 you should be able to determine where the carbonate
species will exhibit the greatest buffering capacity--at pH values of 6.36 and 10.33.
The final expression we need to appreciate is that of the solid-phase. In our
discussion we will use calcite exclusively, but we could certainly substitute for another
solid carbonate phase, such as aragonite or dolomite, in the reactions.
CaCO3 (s) <=> Ca2+ + CO32-
Ksp = 10-8.35
[4.76]
You should already be getting an appreciation of the complexity in the carbonate
system. Due to this complexity, we will consider 4 different possible situations to make
our calculations easier. The 4 possible cases are:
1. A closed system without a solid phase (aqueous phase only)
2. A closed system with a solid phase (aqueous-solid exchange)
3. An open system (gas-aqueous phase exchange) without a solid phase
4. An open system with a solid phase (gas-aqueous-solid phase exchange)
Our objective in each of these systems is to determine the amount of each carbonate
species present. To fulfill this objective we will need to use the equilibrium expressions
presented above (eqns. 4.72-4.76) and introduce 3 additional equations:
First, the dissociation of water:
H2O = H+ + OH-
Kw = 10-14
Second, a mass balance equation for the carbonate species:
{with solids}
2[C]T = [H2CO3*] + [HCO3 ] + [CO3 ] + [CaCO3]
[4.77]
[4.78]
where [CaCO3] would pertain only if calcite were present in the system (cases 2 and 4).
Third, a charge balance (electrical neutrality) equation:
{with solids}
+
[H ] + 2[Ca2+] = [OH-] + [HCO3-] + 2[CO32-]
[4.79]
[Ca2+] would again only enter this equation if calcite were present in the system.
In general, we will always solve complex equilibrium problems with the use of three
types of equations: i) equilibrium, ii) mass balance, and iii) charge balance. Using these
3 equations (4.77-4.79) plus those of the carbonate equilibria (4.72-4.76) we can define
each of the carbonate species for any of the 4 possible systems.
Before we solve each of these 4 systems, it is beneficial to define some
'convenience' factors. The first is that of conditional constants, which provide an easy
means of incorporating non-ideality into complex equilibria.
25
Conditional Constants:
reactions conditions.
Equilibrium constants dependent upon the specific
Conditional constant define the equilibrium only for a single condition. We use these
constants to correct for non-ideal conditions rather than converting our concentrations to
activity. This is convenient in complex equilibria were in obtaining explicitly solutions
we need to combine expressions of equilibria, mass balance, and charge balance. As we
already know, equilibrium equations are based on activities with 'true' equilibrium
constants, but both the mass and charge balance equations are based on concentrations.
Thus, if we switch from equilibrium equation using 'true' constants to 'conditional'
constants we can work all 3 types of equations in concentration units. As an example,
let's look at the first dissociation of H2CO3*.
K1 = (H+)(HCO3-) / (H2CO3*)
Switching from activities to concentrations
K1 = H+[H+] * HCO3-[HCO3-] / [H2CO3*]
K1, conditional = K1 / {H+ * HCO3-} = [H+][HCO3-] / [H2CO3*]
[4.80]
Equation [4.80] defines the condition constant, K1c, for the first dissociation of H2CO3*.
Why is it conditional? Because we divide the 'true' equilibrium constant by the activity
coefficients of the proton and bicarbonate ion, and, of course, the activity coefficients
depend on the specific conditions of the solution. Likewise, we can define the
conditional constants for all the other equilibrium expression in the carbonate system.
K2, conditional = K2 {HCO3-} / {H+} {CO32-}
[4.81]
Kw, conditional = Kw / {H+} {OH-}
[4.82]
Ksp, conditional = Ksp / {Ca2+}{CO32-}
[4.83]
The second convenience factor we will introduce here is the ionization fraction.
Ionization Fraction (): an expression of the potential for dissociation or
protolysis of a weak acid.
This parameter allows one to determine the proportion of each carbonate species in
solution simply by measuring the pH. It is extremely useful for defining the dissociation
26
and thus speciation of any weak acid or base!!! We will make use of this convenience
factor quite a bit throughout the class. We begin with the simplest form of the ionization
fraction, that for a monoprotic acid.
For a monoprotic acid: HA <=> H+ + OH1 = H+ + KHA
[4.84]
[4.86]
(HA)
(H+ )
=
[C]t
( )
[4.87]
(A
– ) = (KHA)
[C] t
( )
[4.88]
Equations 4.86 and 4.87 define the quantity of HA and A- as a function of one
variable, pH, if we know the total amount of acid introduced to the system. Take a close
look at these 3 equations and compare them to the more complex reactions we will
consider next.
Next we move to a diprotic acid, using carbonic acid as an example.
H2CO3* <=> H+ + HCO3- <=> H+ + CO322 = (H+)2 + (H+)K1 + K1K2
[4.89]
[4.90]
(H
2 CO3) (H2 )
= ( )
[C]t
2
[4.91]
–
(HCO
(H+)K1
3)
[C] t = (2)
[4.92]
(CO
2–
K K
3 )
= 1 2
[C] t
( 2)
[4.93]
You may already see a few important facts about ; one of the most important being that
the number of terms in is one more than the number of dissociations. Our final
definition is for a triprotic acid.
H3A <=> H+ + HA2- <=> H+ + HA2- <=> H+ + A33 = (H+)3 + (H+)2K1 + (H+)K1K2 + K1K2K3
(H
3 A) (H +)3
[C] t = (3)
[4.94]
[4.95]
[4.96]
27
(H
2 A– ) (H +)2 K1
=
[C]t
(3)
[4.97]
(HA
2–) (H+)K1 K2
[C]t =
( 3)
[4.98]
(A
3–) K1 K2 K3
[C] t = ( 3)
[4.99]
The utility of should be much more apparent with the high degree of dissociation.
Again, simply by knowing the total amount of acid introduced to the system we can
define the speciation as a function of pH.
The general formula for is:
a) the number of terms equals n + 1 (n = number of dissociations or
protonations)
b) each term represents a component in the dissociation
c) each component is represented by (i) [H+]m, where m is the number of
possible dissociations left for that component, and (ii) the K values for each dissociation
(association) leading to the formation of this species.
An example may make more sense, so let's consider the carbonic acid
dissociation again.
H2CO3* <=> H+ + HCO3- <=> H+ + CO32Since this is a diprotic acid there are 2 dissociations; thus, the number of terms in will
be
terms = n + 1
=2+1=3
There is 1 term for each component: H2CO3*, HCO3-, and CO32-. Knowing that there
are 3 terms we need to define each one. The first term will represent H2CO3*; there are 2
H+ produced from its dissociation so it would be [H+]2. The second term is that of
HCO3-; it represents the product of 1 dissociation and yet can go through 1 more
dissociation; thus, the term for it would be represented by (H+)K1. The final component,
CO32-, has undergone 2 dissociations and is in a fully dissociated state; it is represented
then by K1K2. Putting these terms together gives us 2:
2 = (H+)2 + (H+)K1 + K1K2
The proportion of each species in the dissociation sequence, relative to the total quantity
of the acid or base introduced to the system, is then represented by its -term divided by
. For the carbonate species,
28
(H
2 CO3) (H2 )
= ( )
[C]t
2
–
(HCO
(H+)K1
3)
=
[C] t
(2)
(CO
2–
K K
3 )
= 1 2
[C] t
( 2)
IV-2.2, Individual Carbonate Systems
Case 1: A closed system without a solid phase (aqueous phase only)
This is the most simplified carbonate system; a solution which is neither in
contact with air nor with a solid carbonate phase. Many groundwaters would fall in this
category. They are termed closed systems since the total carbonate content is fixed.
This system is defined by the following equations.
2 Equilibrium Expressions (expressed as concentrations):
[HCO3-] = [H+] [CO32-] / K2c
[4.100]
[H2CO3*] = [H+]2 [CO32-] / K1c K2c
[4.101]
A Mass Balance Expression:
[Carbonate]T = [H2CO3*] + [HCO3-] + [CO32-]
[4.102]
Substituting equations 4.100 and 4.101 into 4.102 and factoring out the carbonate term,
[C]T = [CO2–
3 ]
[CO2–
3 ]
=
[C]T
{
{ K(H K)
(H +)2
(H+)
+
K1cK2c
K2c +1
[4.103]
K2c
+1
(H+)
[4.104]
1c 2c
+ 2
}
+
}
by dividing through by {K1K2 / K1K2} and rearranging,
[CO32-]/[C]T = K1K2 / 2
[4.105]
29
we obtain the ionization parameter expression for CO32-. Using our knowledge of we
can determine the quantity of other species present.
[HCO3-] / [C]T = [H+]K1c / 2
[4.106]
[H2CO3*] / [C]T = [H+]2 / 2
[4.107]
and
were, again, from eqn. [4.90],
2 = [H+]2 + [H+]K1 + K1K2
If we know the total amount of carbonate in the system and we want to know the solution
speciation, then we can solve for all the components as a function of (H+). If we did not
measure pH we could still ascertain the carbonate species by added a charge balance
expression.
Charge Balance Expression:
[H+] = [OH-] + [HCO3-] + 2[CO32-]
[4.108]
then, substituting in the expressions for each species,
[H+] = Kwc / [H+] + [C]T*{ [H+]K1c + 2K1cK2c / 2}
[4.109]
0 = [H+] - Kwc / [H+] - [C]T*{ [H+]K1c + 2K1cK2c / 2}
[4.110]
This is an equation with 1 variable, [H+], which can be solved numerically. After [H+] is
obtained, then all other constituents can be determined simply by knowing the total
carbonate in the system.
Case 2: A closed system with a solid phase (aqueous-solid exchange)
Here, calcite would be in contact with the solution, but gas exchange would be
restricted by some type of barrier. Such a system is again closed because exchange with
the outside atmosphere is restricted and the total carbonate content fixed. Since their is
no gaseous input of carbon dioxide, all of the carbonate species must arise from the
dissolution of calcite (or aragonite). Thus,
[Ca2+] = [C]T = [H2CO3*] + [HCO3-] + [CO32-]
[4.111]
This gives us 6 equations and 6 unknowns (CO32-, HCO3-, H2CO3*, H+, OH-, and Ca2+).
30
With eqn. 4.111 we can define the carbonate species as a function of total
carbonate or calcium since they are equal.
[H2CO3*] = [C]T * ([H+]2 / 2)
[4.112]
[HCO3-] = [C]T * (K1c[H+] / 2)
[4.113]
[CO32-] = [C]T * (K1cK2c / 2)
[4.114]
substituting eqn. 4.114 into the solubility expression, [Ca2+] and [C]T can be determined:
Ksp = (Ca2+) (CO32-) = 10-8.33
[Ca2+] = [C]T = {Ksp,c 2 / K1cK2c}1/2
[4.115]
Case 3: An open system (gas-aqueous phase exchange) without a solid phase
This case represents a 'surface' water, such as a lake, pond, stream, or soil
solution, that is exposed to the atmosphere. Because of the gaseous exchange (open
system) we can not use a mass balance expression. We can, however, use a charge
balance equation;
[H+] = [OH-] + [HCO3-] + 2[CO32-]
[4.116]
We can also develop an expression for each component in this equation.
Equilibrium Expressions:
[H2CO3*] = PCO2 KH
K1c =
[H+] [HCO–3 ]
[H2 CO3 ]
substituting in the expression for H2CO3*
[H+] [HCO–3]
K1c = P
CO 2 KH
[4.117]
rearranging for HCO3--
[HCO–3] =
K1c PCO2 KH
[H+]
[4.118]
31
K2c =
[H+] [CO 2–
3 ]
–
[HCO 3]
[CO2–
3 ] =
=
K2c[HCO–3]
[H+]
K1c K2c PCO2 KH
[H+]2
[4.119]
[4.120]
Kw,c = [H+] [OH-]
[OH–] =
Kw,c
[H+]
[4.121]
Substituting the equilibrium expressions into that of the mass balance leads to,
[H+]3 - {K1c PCO2 KH + Kw,c}[H+] - 2{K1c K2c PCO2KH} = 0
[4.122]
This equation states that if the partial pressure of CO2 is known than the pH can be
determined, followed by each of the carbonate species. Additionally, if the pH were
measured the carbonate species could be determined as well as the partial pressure of
CO2. Solving this set of equations with atmospheric carbon dioxide levels ( PCO2 = 103.5 atm) indicates that the solution will have a pH = 5.6. This in deed is observed.
Distilled water that is in equilibrium with air will not have a pH of 7.0, but rather will be
at pH 5.6 due to the influence of the carbonate system.
Case 4: An open system with a solid phase (gas-aqueous-solid phase exchange)
This obviously the most complicated system we have considered but represents a
very common situation: a surface water exposed to calcite. We will add the dissolution
equation for calcite to the equilibrium expression shown previously, and we will modify
the charge balance expression to include calcium.
Ksp,c = [Ca2+] [CO32-]
The charge balance is now,
[H+] + 2 [Ca2+] = [OH-] + [HCO3-] + 2 [CO32-]
[4.123]
To get an expression in terms of calcium, we can rearrange the solubility expression and
substitute the equation for [CO32-].
32
[Ca 2+] =
=
Ksp,c
[4.124]
[CO2–
3 ]
Ksp,c [H+]2
K1c K2c PCO2 KH
[4.125]
Now, all of the species shown in eqn. 4.123 can be introduced to develop our working
expression:
0=
–
{K
{K
2 Ksp,c
1c
K2c PCO2 KH
1,cPCO2
}[H ]
+ 4
}
+
KH + Kw,c [H ] –
+ [H+] 3
{2K
1c K 2c PCO 2
}
[4.126]
KH
This equation has both [H+] and PCO2 as variables, to solve this equation we must specify
either pH or PCO2. A plot of this equation with pH and PCO2 leads to the Johnstone
Diagram. The Johnstone Diagram is created by specifing a PCO2 level and solving the
equation; then specifying a new PCO2 and solving again until the range of desired values
is covered. The Johnstone Diagram gives a lot of important information. Know how to
use this graph.