NMR Seminar - Powers Group
advertisement

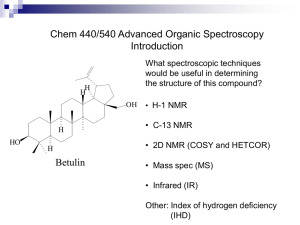
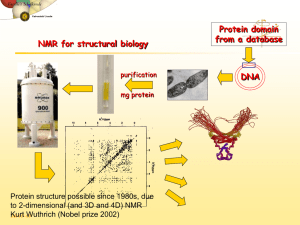
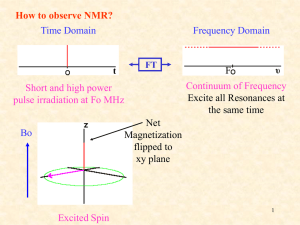
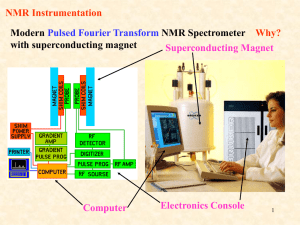
Nuclear Magnetic Resonance A.) Introduction: Nuclear Magnetic Resonance (NMR) measures the absorption of electromagnetic radiation in the radio-frequency region (~4-900 MHz) - nuclei (instead of outer electrons) are involved in absorption process - sample needs to be placed in magnetic field to cause different energy states NMR was first experimentally observed by Bloch and Purcell in 1946 (received Nobel Prize in 1952) and quickly became commercially available and widely used. Probe the Composition, Structure, Dynamics and Function of the Complete Range of Chemical Entities: from small organic molecules to large molecular weight polymers and proteins. NMR is routinely and widely used as the preferred technique to rapidly elucidate the chemical structure of most organic compounds. One of the MOST Routinely used Analytical Techniques Some Suggested NMR References “Spin Dynamics – Basics of Nuclear Magnetic Resonance” M. H. Levitt “Tables of Spectral Data for Structure Determination of Organic Compounds” Pretsch, Clerc, Seibl and Simon “Spectrometric Identification of Organic Compounds” Silverstein, Bassler and Morrill “Organic Structure Determination Using 2-D NMR Spectroscopy: A Problem-Based Approach” Jeffrey H. Simpson “Essential Practical NMR for Organic Chemistry” Stephen A. Richards & John C. Hollerton “Basic One- and Two-Dimensional NMR Spectroscopy” Horst Friebolin “Modern NMR Techniques for Chemistry Research” Andrew E. Derome “Nuclear Magnetic Resonance Spectroscopy” R. K Harris “Experimental Pulse NMR. A Nuts and Bolts Approach” Eiichi Fukushima & Steve B.W. Roeder “Two-Dimensional Nuclear Magnetic Resonance in Liquids”, Ad Bax Some NMR Web Sites The Basics of NMR by J.P. Hornak Hypertext based NMR course http://www.cis.rit.edu/htbooks/nmr/nmr-main.htm Spectral DataBase for Organic Compounds (SDBS) http://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi Educational NMR Software All kinds of NMR software http://deane.bio.ucalgary.ca/EduNMRSoft.html NMR Knowledge Base http://www.spectroscopynow.com/ A lot of useful NMR links NMR Information Server http://www.spincore.com/nmrinfo/ News, Links, Conferences, Jobs Technical Tidbits shimming http://www.acornnmr.com/nmr_topics.htm Useful source for the art of NMR Wiki Sharing NMR know-how http://nmrwiki.org/wiki/index.php?title=Main_Page Some More NMR Web Sites Structure Determination Using NMR by H. J. Reich Web based NMR course http://www.chem.wisc.edu/areas/reich/chem605/index.htm eNMR NMR Periodic Table http://www.bruker-nmr.de/guide/eNMR/chem/NMRnuclei.html Web Spectra http://www.chem.ucla.edu/~webspectra/ Example NMR Structure Problems Organic Structure Elucidation Another Work Book of Unknowns http://www3.nd.edu/~smithgrp/structure/workbook.html NMRShiftDB2 Predict NMR Chemical Shifts http://nmrshiftdb.nmr.uni-koeln.de/nmrshiftdb ChemDoodle Simulate NMR and MS Spectra http://web.chemdoodle.com/demos/simulate-nmr-and-ms NESG Wiki Another NMR Wiki Page, Emphasis in Protein NMR http://www.nmr2.buffalo.edu/nesg.wiki/Main_Page NMR History 1937 1946 1953 1966 1975 1985 Rabi predicts and observes nuclear magnetic resonance Bloch, Purcell first nuclear magnetic resonance of bulk sample Overhauser NOE (nuclear Overhauser effect) Ernst, Anderson Fourier transform NMR Jeener, Ernst 2D NMR Wüthrich first solution structure of a small protein (BPTI) from NOE derived distance restraints 1987 3D NMR + 13C, 15N isotope labeling of recombinant proteins (resolution) 1990 pulsed field gradients (artifact suppression) 1996/7 new long range structural parameters: - residual dipolar couplings from partial alignment in liquid crystalline media - projection angle restraints from cross-correlated relaxation TROSY (molecular weight > 100 kDa) Nobel prizes 1944 Physics Rabi (Columbia) 1952 Physics Bloch (Stanford), Purcell (Harvard) 1991 Chemistry Ernst (ETH) 2002 Chemistry Wüthrich (ETH) 2003 Medicine Lauterbur (University of Illinois in Urbana ), Mansfield (University of Nottingham) NMR History First NMR Spectra on Water 1H NMR spectra of water Bloch, F.; Hansen, W. W.; Packard, M. The nuclear induction experiment. Physical Review (1946), 70 474-85. NMR History First Observation of the Chemical Shift 1H NMR spectra ethanol Modern ethanol spectra Arnold, J.T., S.S. Dharmatti, and M.E. Packard, J. Chem. Phys., 1951. 19: p. 507. O Typical Applications of NMR: 1.) Structural (chemical) elucidation > Natural product chemistry > Synthetic organic chemistry - analytical tool of choice of synthetic chemists - used in conjunction with MS and IR 2.) Study of dynamic processes > reaction kinetics > study of equilibrium (chemical or structural) 3.) Structural (three-dimensional) studies > Proteins, Protein-ligand complexes > DNA, RNA, Protein/DNA complexes > Polysaccharides 4.) Drug Design > Structure Activity Relationships by NMR 5) Medicine -MRI MRI images of the Human Brain O O NH O O OH O OH HO O O O O O Taxol (natural product) NMR Structure of MMP-13 complexed to a ligand Each NMR Observable Nuclei Yields a Peak in the Spectra “fingerprint” of the structure 2-phenyl-1,3-dioxep-5-ene 1H NMR spectra 13C NMR spectra Protein Structures from NMR 2D NOESY Spectra at 900 MHz Lysozyme Ribbon Diagram Information in a NMR Spectra Observable Name Quantitative Information d(ppm) = uobs –uref/uref (Hz) chemical (electronic) environment of nucleus peak separation (intensity ratios) neighboring nuclei (torsion angles) Peak position Chemical shifts (d) Peak Splitting Coupling Constant (J) Hz Peak Intensity Integral unitless (ratio) relative height of integral curve nuclear count (ratio) T1 dependent Peak Shape Line width Du = 1/pT2 peak half-height molecular motion chemical exchange uncertainty principal uncertainty in energy A Basic Concept in ElectroMagnetic Theory A Direct Application to NMR A perpendicular external magnetic field will induce an electric current in a closed loop An electric current in a closed loop will create a perpendicular magnetic field Theory of NMR Quantum Description Nuclear Spin (think electron spin) l a) Nucleus rotates about its axis (spin) a) Nuclei with spin have angular momentum (p) 1) quantized, spin quantum number I 2) 2I + 1 states: I, I-1, I-2, …, -I 3) identical energies in absence of external magnetic field c) NMR “active” Nuclear Spin (I) = ½: 1H, 13C, 15N, 19F, 31P Odd atomic mass I = +½ & -½ NMR “inactive” Nuclear Spin (I) = 0: 12C, 16O Even atomic mass & number Quadrupole Nuclei Nuclear Spin (I) > ½: 14N, 2H, 10B Even atomic mass & odd number I = +1, 0 & -1 Magnetic Moment (m) a) spinning charged nucleus creates a magnetic field Magnetic moment Similar to magnetic field created by electric current flowing in a coil b) magnetic moment (m) is created along axis of the nuclear spin m = gp where: p – angular momentum g – gyromagnetic ratio (different value for each type of nucleus) c) magnetic moment is quantized (m) m = I, I-1, I-2, …, -I for common nuclei of interest: m = +½ & -½ Magnetic alignment = g h / 4p Bo In the absence of external field, each nuclei is energetically degenerate Add a strong external field (Bo). and the nuclear magnetic moment: aligns with (low energy) against (high-energy) Energy Levels in a Magnetic Field a) Zeeman Effect -Magnetic moments are oriented in one of two directions in magnetic field b) Difference in energy between the two states is given by: DE = g h Bo / 2p where: Bo – external magnetic field units:Tesla (Kg s-2 A-1) h – Planck’s constant 6.6260 x 10-34 Js g – gyromagnetic ratio unique value per nucleus 1H: 26.7519 x 107 rad T-1 s2p (observed NMR frequency) c) Frequency of absorption: n = g Bo / d) From Boltzmann equation: Nj/No = exp(-ghBo/2pkT) Energy Levels in a Magnetic Field • Transition from the low energy to high energy spin state occurs through an absorption of a photon of radio-frequency (RF) energy RF Frequency of absorption: n = g Bo / 2p NMR Theory: Classical Description Spinning particle precesses around an applied magnetic field a) Angular velocity of this motion is given by: wo = gBo where the frequency of precession or Larmor frequency is: n = gBo/2p Same as quantum mechanical description Net Magnetization in a Magnetic Field z z Classic View: - Nuclei either align with or against external magnetic field along the z-axis. - Since more nuclei align with field, net magnetization (Mo) exists parallel to external magnetic field Mo x y x y Bo Bo Quantum Description: - Nuclei either populate low energy (a, aligned with field) or high energy (b, aligned against field) - Net population in a energy level. - Absorption of radiofrequency promotes nuclear spins from a b. b DE = h n Bo > 0 a Bo An NMR Experiment We have a net magnetization precessing about Bo at a frequency of wo with a net population difference between aligned and unaligned spins. z z Mo x y x y Bo Bo Now What? Perturbed the spin population or perform spin gymnastics Basic principal of NMR experiments The Basic 1D NMR Experiment Experimental details will effect the NMR spectra and the corresponding interpretation An NMR Experiment resonant condition: frequency (w1) of B1 matches Larmor frequency (wo) energy is absorbed and population of a and b states are perturbed. z Mo B1 w1 z x B1 off… x Mxy (or off-resonance) y y w1 And/Or: Mo now precesses about B1 (similar to Bo) for as long as the B1 field is applied. Again, keep in mind that individual spins flipped up or down (a single quanta), but Mo can have a continuous variation. Right-hand rule Classical Description • Observe NMR Signal Need to perturb system from equilibrium. Net magnetization (Mo) now precesses about Bo and B1 B1 field (radio frequency pulse) with gBo/2p frequency MX and MY are non-zero Mx and MY rotate at Larmor frequency System absorbs energy with transitions between aligned and unaligned states Precession about B1stops when B1 is turned off Mz RF pulse B1 field perpendicular to B0 Mxy Absorption of RF Energy or NMR RF Pulse z Classic View: 90o pulse - Apply a radio-frequency (RF) pulse a long the y-axis - RF pulse viewed as a second field (B1), that the net magnetization (Mo) will precess about with an angular velocity of w1 -- z Mo B1 w1 x B1 off… x Mxy (or off-resonance) y w1 = gB1 precession stops when B1 turned off y w1 b Quantum Description: - enough RF energy has been absorbed, such that the population in a/b are now equal - No net magnetization along the z-axis DE = h n a Bo > 0 Please Note: A whole variety of pulse widths are possible, not quantized dealing with bulk magnetization An NMR Experiment What Happens Next? The B1 field is turned off and Mxy continues to precess about Bo at frequency wo. z x Mxy wo y Receiver coil (x) NMR signal FID – Free Induction Decay Mxy is precessing about z-axis in the x-y plane y Time (s) y y An NMR Experiment The oscillation of Mxy generates a fluctuating magnetic field which can be used to generate a current in a receiver coil to detect the NMR signal. A magnetic field perpendicular to a circular loop will induce a current in the loop. NMR Probe (antenna) NMR Signal Detection - FID The FID reflects the change in the magnitude of Mxy as the signal is changing relative to the receiver along the y-axis Detect signal along X RF pulse along Y Again, the signal is precessing about Bo at its Larmor Frequency (wo). NMR Signal Detection - Fourier Transform So, the NMR signal is collected in the Time - domain But, we prefer the frequency domain. Fourier Transform is a mathematical procedure that transforms time domain data into frequency domain NMR Signal Detection - Fourier Transform After the NMR Signal is Generated and the B1 Field is Removed, the Net Magnetization Will Relax Back to Equilibrium Aligned Along the Z-axis T2 relaxation Two types of relaxation processes, one in the x,y plane and one along the z-axis Peak shape also affected by magnetic field homogeneity or shimming NMR Relaxation a) No spontaneous reemission of photons to relax down to ground state Probability too low cube of the frequency b) Two types of NMR relaxation processes spin-lattice or longitudinal relaxation (T1) i. transfer of energy to the lattice or solvent material ii. coupling of nuclei magnetic field with magnetic fields created by the ensemble of vibrational and rotational motion of the lattice or solvent. iii. results in a minimal temperature increase in sample Mz = M0(1-exp(-t/T1)) Recycle Delay: General practice is to wait 5xT1 for the system to have fully relaxed. NMR Relaxation spin-spin or transverse relaxation (T2) i. exchange of energy between excited nucleus and low energy state nucleus ii. randomization of spins or magnetic moment in x,y-plane iii. related to NMR peak line-width Mx = My = M0 exp(-t/T2) (derived from Heisenberg uncertainty principal) Please Note: Line shape is also affected by the magnetic fields homogeneity NMR Sensitivity The applied magnetic field causes an energy difference between aligned(a) and unaligned(b) nuclei b Low energy gap DE = h n Bo > 0 a Bo = 0 The population (N) difference can be determined from Boltzmman distribution: Na / Nb = e DE / kT The DE for 1H at 400 MHz (Bo = 9.5 T) is 3.8 x 10-5 Kcal / mol Na / Nb = 1.000064 Very Small ! ~64 excess spins per million in lower state NMR Sensitivity NMR signal depends on: signal (s) ~ g4Bo2NB1g(u)/T 1) Number of Nuclei (N) (limited to field homogeneity and filling factor) 2) Gyromagnetic ratio (in practice g3) 3) Inversely to temperature (T) 4) External magnetic field (Bo2/3, in practice, homogeneity) 5) B12 exciting field strength DE = g h Bo / 2p Na / Nb = e DE / kT Increase energy gap -> Increase population difference -> Increase NMR signal DE ≡ Bo ≡ g - Intrinsic property of nucleus can not be changed. g NMR Sensitivity • Relative sensitivity of 1H, 13C, 15N and other nuclei NMR spectra depend on Gyromagnetic ratio (g) Natural abundance of the isotope g - Intrinsic property of nucleus can not be changed. (gH/gC)3 1H for 13C is 64x (gH/gN)3 for 15N is 1000x is ~ 64x as sensitive as 13C and 1000x as sensitive as 15N ! Consider that the natural abundance of 13C is 1.1% and 15N is 0.37% relative sensitivity increases to ~6,400x and ~2.7x105x !! 1H NMR spectra of caffeine 8 scans ~12 secs 13C NMR spectra of caffeine 8 scans ~12 secs 13C NMR spectra of caffeine 10,000 scans ~4.2 hours NMR Sensitivity Increase in Magnet Strength is a Major Means to Increase Sensitivity NMR Sensitivity But at a significant cost! ~$800,000 ~$2,000,000 ~$4,500,000 Chemical Shift Up to this point, we have been treating nuclei in general terms. Simply comparing 1H, 13C, 15N etc. If all 1H resonate at 500MHz at a field strength of 11.7T, NMR would not be very interesting The chemical environment for each nuclei results in a unique local magnetic field (Bloc) for each nuclei: Beff = Bo - Bloc --- Beff = Bo( 1 - s ) s is the magnetic shielding of the nucleus Chemical Shift a) Small local magnetic fields (Bloc) are generated by electrons as they circulate nuclei. • Current in a circular coil generates a magnetic field b) These local magnetic fields can either oppose or augment the external magnetic field • Typically oppose external magnetic field • Nuclei “see” an effective magnetic field (Beff) smaller then the external field • s – magnetic shielding or screening constant i. depends on electron density ii. depends on the structure of the compound Beff = Bo - Bloc --- Beff = Bo( 1 - s ) Chemical Shift Beff = Bo - Bloc --- Beff = Bo( 1 - s ) HO-CH2-CH3 s – reason why observe three distinct NMR peaks instead of one based on strength of B0 n = gBo/2p de-shielding high shielding Shielding – local field opposes Bo Chemical Shift Effect of Magnetic Anisotropy 1) external field induces a flow (current) of electrons in p system – ring current effect 2) ring current induces a local magnetic field with shielding (decreased chemical shift) and deshielding (increased chemical shifts) Decrease in chemical shifts Increase in chemical shifts The NMR scale (d, ppm) Bo >> Bloc -- MHz compared to Hz Comparing small changes in the context of a large number is cumbersome d= n - nref nref ppm (parts per million) Instead use a relative scale, and refer all signals (n) in the spectrum to the signal of a particular compound (nref ). IMPORTANT: absolute frequency is field dependent (n = g Bo / 2p) CH 3 Tetramethyl silane (TMS) is a common reference chemical H3C Si CH 3 CH 3 The NMR scale (d, ppm) Chemical shift (d) is a relative scale so it is independent of Bo. Same chemical shift at 100 MHz vs. 900 MHz magnet IMPORTANT: absolute frequency is field dependent (n = g Bo / 2p) At higher magnetic fields an NMR spectra will exhibit the same chemical shifts but with higher resolution because of the higher frequency range. NMR Spectra Terminology TMS CHCl3 7.27 increasing d low field down field high frequency (u) de-shielding Paramagnetic 600 MHz 1H 0 decreasing d high field up field low frequency high shielding diamagnetic 150 MHz 13C ppm 92 MHz 2H Increasing field (Bo) Increasing frequency (u) Increasing g Increasing energy (E, consistent with UV/IR) Chemical Shift Trends For protons, ~ 15 ppm: For carbon, ~ 220 ppm: Carbon chemical shifts have similar trends, but over a larger sweep-width range (0-200 ppm) Chemical Shift Trends Acids Aldehydes Alcohols, protons a to ketones Aromatics Amides Olefins Aliphatic ppm 15 C=O in ketones 10 7 5 Aromatics, conjugated alkenes Olefins 2 0 TMS Aliphatic CH3, CH2, CH ppm 210 150 C=O of Acids, aldehydes, esters 100 80 50 0 TMS Carbons adjacent to alcohols, ketones CHARACTERISTIC PROTON CHEMICAL SHIFTS Common Chemical Shift Ranges Carbon chemical shifts have similar trends, but over a larger sweep-width range (0-200 ppm) Type of Proton Structure Chemical Shift, ppm Cyclopropane C3H6 0.2 Primary R-CH3 0.9 Secondary R2-CH2 1.3 Tertiary R3-C-H 1.5 Vinylic C=C-H 4.6-5.9 Acetylenic triple bond,CC-H 2-3 Aromatic Ar-H 6-8.5 Benzylic Ar-C-H 2.2-3 Allylic C=C-CH3 1.7 Fluorides H-C-F 4-4.5 Chlorides H-C-Cl 3-4 Bromides H-C-Br 2.5-4 Iodides H-C-I 2-4 Alcohols H-C-OH 3.4-4 Ethers H-C-OR 3.3-4 Esters RCOO-C-H 3.7-4.1 Esters H-C-COOR 2-2.2 Acids H-C-COOH 2-2.6 Carbonyl Compounds H-C-C=O 2-2.7 Aldehydic R-(H-)C=O 9-10 Hydroxylic R-C-OH 1-5.5 Phenolic Ar-OH 4-12 Enolic C=C-OH 15-17 Carboxylic RCOOH 10.5-12 Amino RNH2 1-5 Predicting Chemical Shift Assignments Numerous Experimental NMR Data has been compiled and general trends identified • See: “Tables of Spectral Data for Structure Determination of Organic Compounds” Pretsch, Clerc, Seibl and Simon “Spectrometric Identification of Organic Compounds” Silverstein, Bassler and Morrill • Spectral Databases: Aldrich/ACD Library of FT NMR Spectra Sadtler/Spectroscopy (UV/Vis, IR, MS, GC and NMR) http://www.chem.wisc.edu/areas/reich/chem605/index.htm Ongoing effort to predict chemical shifts from first principals (quantum mechanical description of factors contributing to chemical shifts) See: Cynthia J. Jameson, “Understanding NMR Chemical Shifts”, Annu. Rev. Phys. Chem. 1996. 47:135–69 Predicting Chemical Shift Assignments Empirical Chemical Shift Trends (Databases) Have Been Incorporated Into A Variety of Software Applications Example: ChemDraw • Program that allows you to generate a 2D sketch of any compound • can also predict 1H and 13C chemical shifts matches sub-fragments of structure to structures in database Fulvene Protocol of the H-1 NMR Prediction: 5.22 H H5.22 Node Shift Base + Inc. H 6.44 H 6.44 H 6.44 H 6.44 H 5.22 H 5.22 5.25 1.24 -0.05 5.25 -0.05 1.24 5.25 1.24 -0.05 5.25 -0.05 1.24 5.25 -0.03 5.25 -0.03 6.44 H H6.44 H 6.44 H 6.44 Estimation Quality: blue = good, magenta = medium, red = rough 6 5 4 PPM 3 Comment (ppm rel. to TMS) 1-ethylene 1 -C=C gem 1 -C=C trans 1-ethylene 1 -C=C trans 1 -C=C gem 1-ethylene 1 -C=C gem 1 -C=C trans 1-ethylene 1 -C=C trans 1 -C=C gem 1-ethylene 2 -C=C c + t 1-ethylene 2 -C=C c + t 2 1 0 Predicting Chemical Shift Assignments How Does the Predicted Results Compare to Experimental Data? Parameter D(A) D(B) D(C) Experimental ( ppm) 6.22 6.53 5.85 Predicted (ppm) 6.44 6.44 5.22 Typical accuracy A number of factors can affect prediction: Similarity of structures in reference database Solvent Temperature structure/conformation additive nature of parts towards the whole Predicting Chemical Shift Assignments Experimental NMR Data has also been used to Develop Web-Based Tools to Predict NMR Spectra Example: nmrdb.org NMR Predictor (http://www.nmrdb.org/new_predictor) • Program that allows you to generate a 2D sketch of any compound • Predicts 1H chemical shifts Demo of ChemDraw Coupling Constants through-bond interaction that results in the splitting of a single peak into multiple peaks of various intensities 1) The spacing in hertz (hz) between the peaks is a constant i. coupling constant (J) bonding electrons convey spin states of bonded nuclei 1) spin states of nuclei are “coupled” 2) alignment of spin states of bonded nuclei affects energy of the ground (a) and excited states (b) of observed nuclei 3) Coupling pattern and intensity follows Pascal’s triangle Coupling Constants Energy level of a nuclei are affected by covalently-bonded neighbors spin-states 1 H 13 1 1 H H three-bond C one-bond Spin-States of covalently-bonded nuclei want to be aligned. +J/4 I -J/4 bb S ab J (Hz) ba S +J/4 I aa I S The magnitude of the separation is called coupling constant (J) and has units of Hz. Coupling Constants 1 11 121 1331 14641 1 5 10 10 5 1 1 6 15 20 15 6 1 1 7 21 35 35 21 7 1 Common NMR Splitting Patterns Multiplets consist of 2nI + 1 lines I is the nuclear spin quantum number (usually 1/2) and n is the number of neighboring spins. singlet doublet triplet quartet 1:1 1:2:1 1:3:3:1 pentet 1:4:6:4:1 Coupling Rules: 1. 2. 3. 4. 5. 6. equivalent nuclei do not interact coupling constants decreases with separation ( typically # 3 bonds) multiplicity given by number of attached equivalent protons (n+1) multiple spin systems multiplicity (na+1)(nb+1) Relative peak heights/area follows Pascal’s triangle Coupling constant are independent of applied field strength IMPORTANT: Coupling constant pattern allow for the identification of bonded nuclei. Karplus Equation – Coupling Constants J = const. + 10Cosf Relates coupling constant to Torsional angle. Used to solve Structures! Example: The proton NMR spectrum is for a compound of empirical formula C4H8O. Identify the compound Absence of peak at ~9.7 ppm eliminates aldehyde group And suggests ketone Triplet at ~1.2 ppm suggests a methyl group coupled to a methylene group Strong singlet at ~2.25 ppm methyl next to carbonyl Quartet at ~2.5 ppm suggests a methylene next to a carbonyl coupled to a methyl Nuclear Overhauser Effect (NOE) a) b) c) Interaction between nuclear spins mediated through empty space (#5Å) like ordinary bar magnets Important: effect is time-averaged Gives rise to dipolar relaxation (T1 and T2) and specially to cross-relaxation Perturb 1H spin population affects 13C spin population NOE effect Nuclear Overhauser Effect (NOE) Nuclear Overhauser Effect (NOE, h) – the change in intensity of an NMR resonance when the transition of another are perturbed, usually by saturation. hi = (I-Io)/Io where Io is thermal equilibrium intensity Saturation – elimination of a population difference between transitions (irradiating one transition with a weak RF field) irradiate bb ab N N-d X A ba X aa N+d N A Populations and energy levels of a homonuclear AX system (large chemical shift difference) Observed signals only occur from single-quantum transitions Nuclear Overhauser Effect (NOE) Saturated (equal population) ab N-½d saturate bb N-½d I S ba I aa N+½d N+½d S Saturated (equal population) Observed signals only occur from single-quantum transitions Populations and energy levels immediately following saturation of the S transitions bb ab W1A N-½d W1X N-½d W2 W0 aa W1X N+½d ba W1A Relaxation back to equilibrium can occur through: Zero-quantum transitions (W0) Single quantum transitions (W1) Double quantum transitions (W2) N+½d The observed NOE will depend on the “rate” of these relaxation pathways Nuclear Overhauser Effect (NOE) Mechanism for Relaxation • Dipolar coupling between nuclei local field at one nucleus is due to the presence of the other – depends on orientation of the whole molecule • Dipolar coupling, T1 and NOE are related through rotational correlation time (tc) – rotational correlation is the time it takes a molecule to rotate one radian (360o/2p). • Relaxation or energy transfers only occurs if some frequencies of motion match the frequency of the energy of transition – the available frequencies for a molecule undergoing Brownian tumbling depends on tc – W1A 3t c 3t c r 6 (1 w A2t c2 ) r6 3t c 2t c W0 6 6 2 2 r (w A w X ) t c ) r W2 12t c 12t c r 6 (1 (w A w X ) 2t c2 ) r6 NOE is dependent on the distance (1/r6) separating the two dipole coupled nuclei Important: the effect is time-averaged! 2D NOESY (Nuclear Overhauser Effect) Relative magnitude of the cross-peak is related to the distance (1/r6) between the protons (≥ 5Ǻ). NOE is a relaxation factor that builds-up during The “mixing-time (tm) NMR Structure Determination NOE Data Is the Fundamental Piece of Information to Determine Any Structure (DNA, RNA, Protein, small molecule) 2D NOESY Spectra at 900 MHz Lysozyme Ribbon Diagram NMR Instrumentation (block diagram) Superconducting Magnet a) solenoid wound from superconducting niobium/tin or niobium/titanium wire b) kept at liquid helium temperature (4K), outer liquid N2 dewar near zero resistance minimal current lose magnet stays at field for years without external power source Cross-section of magnet magnet spinner sample lift NMR Tube RF coils cryoshims shimcoils Superconducting solenoid Use up to 190 miles of wire! Probe Liquid N2 Liquid He Superconducting Magnet • Problems: − Field drifts (B0 changes) Field Drift over 11 Hrs (~ 0.15Hz/hr Remember: n = gBo/2p Lock System a) c) Corrects for magnetic field drift NMR probes contains an additional transmitter coil tuned to deuterium frequency changes in the intensity of the reference absorption signal controls a feedback circuit; a frequency generator provides a fixed reference frequency for the lock signal Lock Feedback Circuit if the observed lock signal differs from the reference frequency, a small current change occurs in a room-temperature shim coil (Z0) to create a small magnetic field to augment the main field to place the lock-signal back into resonance Lock Changes From Off-resonance to Onresonance Superconducting Magnet • Problems: − Field is not constant over sample (spatial variation) Again: n = gBo/2p Magnetic Field Homogeneity Frequency of absorption: Poor Homogeneity multiple peaks at different effective Bo Resonance depends on position in NMR sample n = g Bo / 2p Good Homogeneity single peak with frequency dependent on Bo Shim System • • Corrects for magnetic inhomogeneity Spatial arrangement of 20 or more coils actual shim coils Sketch of shim coils change current in each coil to “patch” differences in field and fix distortions in peak shape Shim Coils • Optimize shims by i) minimizing line-width, ii) maximizing lock signal or iii) maximizing FID • Examples of poor line-shapes due to shimming errors Tune and Match System a) c) d) Tune- corrects the differences between observed and desired frequency Match – correct impedance difference between resonant circuit and transmission line (should be 50W ) Power submitted to transmitter and receiver is maximized Adjust two capacitors until the tuning and desired frequency match and you obtain a null Affects: signal-to-noise accuracy of 90o pulse sample heating chemical shift accuracy Receiver Gain a) b) c) Amplifies the radio frequency FID signal from the probe Set to maximize signal-to-noise Signal is dependent on sample concentration The Analog to Digital Converter (ADC) has a maximum range of integers Clipped Fid Clipped FID Dynamic Range Problem Continuous Wave (CW) vs. Pulse/Fourier Transform NMR Sensitivity Issue A frequency sweep (CW) to identify resonance is very slow (1-10 min.) Step through each individual frequency. Pulsed/FT collect all frequencies at once in time domain, fast (N x 1-10 sec) NMR Pulse a) In FT-NMR, how are all the individual nuclei excited simultaneously? b) RF pulses are typically short-duration (msecs) - produces bandwidth (1/4t) centered around single frequency - shorter pulse width broader frequency bandwidth 1H 6 ms 90o pulse ±41666 Hz ±69.4 ppm at 600 MHz Heisenberg Uncertainty Principal: Du.Dt ~ 1/2p A radiofrequency pulse is a combination of a wave (cosine) of frequency w o and a step function * = tp Pulse length (time, tp) FT The Fourier transform indicates the pulse covers a range of frequencies NMR Pulse NMR pulse length or Tip angle (tp) z Mo z x qt tp x B1 Mxy y y qt = g * tp * B1 The length of time the B1 field is on => torque on bulk magnetization (B1) A measured quantity – instrument and sample dependent. NMR Pulse Some useful common pulses z z 90o pulse Mo Maximizes signal in x,y-plane where NMR signal detected x p/2 90o y x Mxy y z 180o pulse Inverts the spin-population. No NMR signal detected Mo z x y Can generate just about any pulse width desired. p 180o x y -Mo NMR Data Detection and Processing Fourier Transform NMR Increase signal-to-noise (S/N) by collecting multiple copies of FID and averaging signal. S / N number of scans But, total experiment time is proportional to the number of scans exp. time ~ (number of scans) x (recycle delay) Proper Spectral Width Needs to be large enough to capture all the NMR resonances Correct Spectra Spectra with carrier offset resulting in peak folding or aliasing Sweep Width (range of radio-frequencies monitored for nuclei absorptions) Quadrature detection a) Frequency of B1 (carrier) is set to the center of the spectra. - Small pulse length to excite the entire spectrum - Minimizes folded noise b) How to differentiate between peaks upfield and downfield from carrier? - observed peak frequencies are all relative to the carrier frequency c) If carrier at edge of spectra, peaks are all positive or negative relative to carrier - Excite twice as much noise, decrease S/N How to differentiate between magnetization that precesses clockwise and counter clockwise? Quadrature detection carrier carrier same frequency relative to the carrier, but opposite sign. Quadrature detection PH = 0 B B PH = 90 Use two detectors 90o out of phase. F w (B1) F PH = 0 F S F S Phase of Peaks are different. PH = 90 Digital Resolution – number of data points The FID is digitized Equal delay between points (dwell time) DT = 1 / (2 * SW) Want to maximize digital resolution, more data points increases acquisition time (AQ) and experimental time (ET): AQ = DT x NP ET = AQ x NS larger spectral width (SW) requires more data points for the same resolution Digital Resolution – number of data points Want to maximize digital resolution: Proper Acquisition Time Needs to be long enough to allow FID to Completely Decay Sinc Wiggles Truncated FID Zero Filling Improve digital resolution by adding zero data points at end of FID 4K data 4K FID No zero-filling 16K zero-fill 20K FID 16K zero-filling Window Functions Emphasize the signal or decrease the noise by applying a mathematical function to the FID. Good stuff Mostly noise Sensitivity Resolution F(t) = 1 * e - ( LB * t ) – line broadening Effectively adds LB in Hz to peak Line-widths Can either increase S/N or Resolution Not Both! LB = 5.0 Hz Increase Sensitivity FT LB = -1.0 Hz Increase Resolution FT Baseline Correction Need a flat baseline – prefer to fix experimentally Apply any number of mathematical functions: linear Polynomial (upwards of 6-orders) FID reconstruction Penalized parametric smoothing Etc. A number of factors lead to baseline distortions: Intense solvent or buffer peaks Phasing problems Errors in first data points of FID Short recycle tines Short acquisition times Receiver gain Xi & Roche BMC Bioinformatics (2008) 9:234 Phase Correction Need a flat baseline – phase all peaks to be pure absorptive in shape Phase is due to difference in actual time and the zero-time point of FID (sin + cosines) Improper phasing can cause severe distortions in the baseline Xi & Roche BMC Bioinformatics (2008) 9:234 NMR Peak Integration or Peak Area a) The relative peak intensity or peak area is proportional to the number of protons associated with the observed peak. b) Means to determine relative concentrations of multiple species present in an NMR sample. Relative peak areas = Number of protons 3 Integral trace HO-CH2-CH3 2 1 Exchange Rates and NMR Time Scale i. Time Scale Slow Intermediate Fast Range (Sec-1) NMR time scale refers to the chemical shift time scale a) remember – frequency units are in Hz (sec-1) time scale b) exchange rate (k) c) differences in chemical shifts between species in exchange indicate the exchange rate. Chem. Shift (d) k << dA- dB k = dA - dB k >> dA - dB 0 – 1000 Coupling Const. (J) k << JA- JB k = JA- JB k >> JA- JB 0 –12 T2 relaxation k << 1/ T2,A- 1/ T2,B k = 1/ T2,A- 1/ T2,B k >> 1/ T2,A- 1/ T2,B 1 - 20 d) For systems in fast exchange, the observed chemical shift is the average of the individual species chemical shifts. dobs = f1d1 + f2d2 f1 +f2 =1 where: f1, f2 – mole fraction of each species d1,d2 – chemical shift of each species ii. Effects of Exchange Rates on NMR data k = p Dno2 /2(he - ho) k = p Dno / 21/2 k = p (Dno2 - Dne2)1/2/21/2 k = p (he-ho) k – exchange rate h – peak-width at half-height n – peak frequency e – with exchange o – no exchange NMR Dynamics and Exchange Equal Population of Exchange Sites No exchange: With exchange: W1/ 2 = 1 1 pT2 pt ex k= 1 t ex slow k = 0.1 s-1 k = 5 s-1 Increasing Exchange Rate W1/ 2 1 = pT2 k = 10 s-1 k = 20 s-1 k = 40 s-1 coalescence k = 88.8 s-1 k = 200 s-1 k = 400 s-1 k = 800 s-1 fast k = 10,000 s-1 40 Hz Multidimensional NMR NMR pulse sequences a) composed of a series of RF pulses, delays, gradient pulses and phases b) in a 1D NMR experiment, the FID acquisition time is the time domain (t1) c) more complex NMR experiments will use multiple “time-dimensional” to obtain data and simplify the analysis. d) Multidimensional NMR experiments may also use multiple nuclei (2D, 13C,15N) in addition to 1H, but usually detect 1H) 1D NMR Pulse Sequence Creating Multiple Dimensions in NMR a) collect a series of FIDS incremented by a second time domain (t1) b) the normal acquisition time is t2. c) Fourier transformation occurs for both t1 and t2, creating a twodimensional (2D) NMR spectra Relative appearance of each NMR spectra will be modulated by the t1 delay Creating Multiple Dimensions in NMR In 2D NMR spectra, diagonal peaks are normal 1D peaks, off-diagonal or crosspeaks indicate a correlation between the two diagonal peaks Collections of FIDs with t1 modulations Fourier Transform t1 obtain 2D NMR spectra Fourier Transform t2 obtain series of NMR spectra modulated by t1 Looking down t1 axis, each point has characteristics of time domain FID Peaks along diagonal are normal 1D NMR spectra Contour map (slice at certain threshold) of 3D representation of 2D NMR spectra. (peak intensity is third dimension Cross-peaks correlate two diagonal peaks by J-coupling or NOE interactions 2D 1H-13C HSQC Experiment Correlates all directly bonded 13C-1H pairs generally requires 13C-labeling (1.1% natural abundance) 2D 1H-1H TOCSY Experiment Correlates all 3-bonded 1H-1H pairs in a molecules