PHARM4515-16 (NSAIDs)
advertisement
")
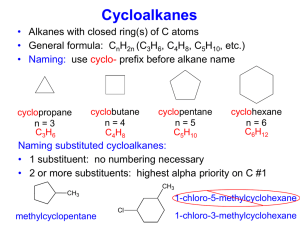
NSAIDs Books: 1. Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry 11th ed. Lippincott, Williams & Wilkins ed. Structurally diverse agents with anti-inflammatory activity Activity is attributed to their ability to inhibit cyclooxygenase (COX) Cyclooxygenase involved in the biosynthesis of prostaglandins Prostaglandins are a class of eicosanoids Eicosanoids are any product derived from arachidonic acid, a twenty carbon fatty acid Eicosanoids also include, thromboxanes, lipoxins, and leukotrienes. Natural Eicosanoids PGA2 O O 9 11 HO 13 O CH3 20 15 OH O COOH COOH CH3 CH3 OH PGE2 HO COOH COOH CH3 CH3 CH3 HO PGG2 HO OH PGH2 COOH CH3 O O COOH COOH CH3 CH3 O OH PGI2 Prostacycline TXA2 Thromboxane A2 O O OH CH3 O OH CH3 OH TXB2 COOH COOH OH PGJ2 O OOH OH PGF2 COOH OH O OH PGC2 OH PGD2 O PGB2 1 COOH 5 HO COOH CH3 O OH Commercial Prostanoids A. Ocular Hypertension & Glaucoma BIMATOPROST HO Lumigan LATANOPROST HO Xalatan N H O CH3 O O HO HO HO OH TRAVOPOST Travatan O O OH CH3 OH UNOPROSTONE isopropyl Rescula HO O HO CH3 CF3 CH3 O O CH3 HO O CH3 CH3 CH3 Commercial Prostanoids -II B. Pulonary Hypertension EPOPROSTENOL PGI Flolan ILOPROST Ventavis ONa COOH COOH TREPROSTINIL sodium Remodulin O OH H O CH3 CH3 O CH3 H HO HO OH OH C. Other Uses CARBOPROST Hemabate O HO COOH MISOPROSTOL Cytotec,(Arthortec) OH CH3 HO H3 C O CH3 CH3 HO OH DINOPROSTONE Prostglandin E2 Prostin E2, Prepidil, Cervidil ALPROSTADIL Prostglandin E1 Prostin VR, Caverject, Edex, Muse O O COOH COOH CH3 CH3 HO CH3 O OH HO OH OH Prostaglandins - Nomenclature Prostanoic acid 9 1 COOH O PGE1 O PGE2 O COOH PGE3 COOH COOH 20 11 15 HO OH HO OH HO OH The nomenclature is derived from the hypothetical compound prostanoic acid The different prostaglandins are divided into several main classes (A, B, C, D, E, F, G, H) depending on the type and spatial relationship of the oxygen functions on C-9 and C-11 of the cyclopentane ring (all have a hydroxyl on C-15) The designation refers to the steroeochemistry of the OH at C-9, below and on the same side (cis) of the ring as the ––OH on C-11. Prostaglandins - Function A class of highly active endogenous mediators Depending on the individual Prostaglandin and the tissue they exert many varied actions Prostaglandins are also implicated in the inflammatory response and in sensitizing pain receptors to the action of other mediators Occurring during acute and chronic inflammatory illness, prostaglandins are produced at the site of inflammation where they mediate many of the symptoms of inflammation such as edema and pain Play critical roles in tissue homeostasis and function They have a cytoprotective role in the kidney and gastric mucosa. Prostaglandins - Biosynthesis P ho s ph o lip ase A 1 P ho s ph o lip ase A 2 O O O C (CH2 )nCH3 C O O O CH3 P N O O CH3 P ho s ph o lip ase C CH3 P ho s ph o lip ase D Prostaglandins are biosynthesized from Arachidonic acid Archidonic acid is found esterified as a cell membrane phospholipid The concentration of free arachidonic acid is low The biosynthesis of the eicosanoids depends primarily on its release from cellular stores by acyl hydrolases or phospolipases. Biosynthesis is enhanced by many physical, chemical and hormonal stimuli and involves activation of enzymes by an increased concentration of calcium Membrane bound Phospholipase A2 is involved in the release of arachidonic acid. Action of cyclooxygenase on arachidonic acid results oxygenated products containing ring structures: prostaglandins, thromboxanes, and prostacyclin action of various lipoxygenases result the hydroxylated products: HPETEs, HETEs, lipoxins and leukotrienes THE EICOSANOIDS The Arachidonic Acid Cascade O O C O C (CH2)nCH3 O O O CH3 P O Lipoxins N O H 3C CH3 Phospholipase A2 [3.1.1.14] Arachinate 15–lipoxygenase [EC 1.13.11.33] 15–HPETE COOH Cytochrome P450 Arachinate 12–lipoxygenase [EC 1.13.11.31] Arachidonic acid Arachinate 5–lipoxygenase [EC 1.13.11.34] Epoxyeicosatriene acids Dihydroxy acids Cyclooxygenase 5–HPETE COOH 12–HPETE O O Leukotrienes LTA4 LTC4 LTB4 COOH O LTD4 O LTF4 COOH O O LTE4 OOH Prostaglandin G2 LT = Leukotriene PG = Prostaglandin TX = Thromboxane HETE = Hydroxyeicosatetraenoic acid HPETE = Hydroperoxyeicosatetraenoic acid Peroxidase COOH O O Prostaglandins OH PGA2 PGB2 PGE2 PGC2 PGF2 PGD2 PGI2 Prostaglandin H2 Thromboxanes TXB2 TXA2 O C H COOH H2C C H O OH Malondialdehyde Hydroxyhepta decatrienoic aacid HHT HO OH COOH COOH HO Arachidonic acid OH Thromboxane B2 1 O O COOH COOH COOH O O OOH Prostaglandin G2 HO OH OH 19–Hydroxy Prostaglandin F2a OH Thromboxane A2 7 2 HO HO COOH HO O 3 O O COOH OH Prostaglandin H2 OH Prostaglandin F2a O O 6 COOH O 5 OH Prostaglandin D2 PGH2 is also converted into two unstable yet highly active thromboxanes (so named because they were first isolated from thrombocytes) 4 COOH COOH COOH O HO OH OH 19–Hydroxy Prostaglandin E2 O HO OH Prostaglandin E2 O HO COOH COOH OH OH 19–Hydroxy Prostaglandin A2 OH Prostaglandin I2 Prostacyclin COOH O O COOH COOH HO OH 6–Keto– Prostaglandin F1a OH Prostaglandin C2 OH OH 19–Hydroxy Prostaglandin C2 O COOH OH Prostaglandin B2 Prostaglandin endoperoxide synthase 2. Peroxidase [EC 1.14.99.1] Prostaglandin endoperoxide synthase (cylcooxyenase domain) Prostaglandin endoperoxide synthase (hydroperoxidase domain) 1. Cyclooxygenase OH Prostaglandin A2 3. 4. 5. 6. 7. Prostaglandin F2a synthase [EC 1.1.1.188] Prostaglandin E2 synthase [EC 5.3.99.3] Prostaglandin I2 synthase [EC 5.3.99.4] Prostaglandin D2 synthase [EC 5.3.99.2] Thromboxane A2 synthase [EC 5.3.99.5] Products of 5–Lipoxygenases Leukotriene Biosynthesis COOH Arachidonic acid 1 OOH OH COOH CH3 inhibited by O Zileuton N HO NH2 S COOH 2 5–HPETE 5–HETE 1 O COOH C5H11 Leukotriene A4 4 3 1. 2. 3. 4. 5. 6. 5–Lipoxygenase + FLAP [EC 1.13.11.34] Peroxidase Leukotriene A4 epoxide hyrolase (LTA4 hydrolase) [EC 3.3.2.6] LTC4 synthetase (a glutathione–S–transferase) [EC 2.5.1.37] g–Glutamyl transferase [EC 2.3.2.2] Cysteinyl glycinase (an amino dipeptidase) HO H COOH C5H11 C5H11 OH Leukotriene B4 5 HO H COOH C5H11 H S CHCOOH NH2 Leukotriene E4 6 HO H COOH S CHCONHCH2COOH NHCOCH2CH2CHCOOH Leukotriene C4 NH 2 COOH C5H11 H S CHCONHCH2COOH NH2 Leukotriene D4 HO H COOH C5H11 H S CHCOOH NCOCH2CH2CHCOOH Leukotriene F4 NH 2 Products of 15–Lipooxygenases Lipoxin Biosynthesis COOH Arachidonic acid 1 COOH COOH 2 OH OOH 15–HPETE 15–HETE 3 OOH OH COOH COOH 2 HPETE – Hydroperoxyeicosatetraenoic acid HETE – Hydroxyeicosatetraenoic acid OOH OH 5,15–HETE 5,15–HPETE COOH O 1 Arachinate 15–lipoxygenase [EC 1.13.11.33] 2 Peroxidase 3 Arachinate 5–Lipoxygenase [EC 1.13.11.34] OH HO OH HO OH COOH COOH OH 6S–Lipoxin A OH OH COOH OH Lipoxin B OH Lipoxin C Protaglandin Endoperoxide Synthase In 1971, John Vane and his colleagues showed that aspirin and other nonsteroidal antiinflammatory drugs inhibited the enzyme, cyclooxygenase and that this inhibition was responsible for their anti-inflammatory properties The biosynthetic reactions are catalyzed by a enzyme complex commonly named Prostaglandin endoperoxide synthase which is located in the endoplasmic reticulum It is a bifunctional enzyme with two catalytic sites adjacent but spatially distinct On one side, it has the cyclooxygenase active site and on the opposite side, it has an entirely separate peroxidase site, which is needed to activate the heme groups that participate in the cyclooxygenase reaction 1. First COX oxidizes and cyclizes arachidonic acid, forming PGG2, which has an endoperoxide as well as an exoperoxide (that gives the name cyclooxygenase) 2. PGG2 then diffuse to the peroxidase catalytic site where the exoperoxide at C15 is reduced to PGH2 3. PGG2 and PGH2 are chemically unstable (t1/2 of 5 min) and are converted enzymatically by enzymes called synthetases into the other prostaglandins 3D Structure of the Enzyme Complex The enzyme complex is a dimer of identical subunits, so altogether, there are two cyclooxygenase active sites and two peroxidase active sites in close proximity Each subunit has a small carbon-rich knob that anchor the complex to the membrane of the endoplasmic reticulum, shown in light blue at the bottom of the picture The cyclooxygenase active site is buried deep within the protein, and is reachable by a tunnel that opens out in the middle of the knob. This acts like a funnel, guiding arachidonic acid out of the membrane and into the enzyme for processing COX-1 & COX-2 Side pocket In COX1 residues Arg120 &Tyr355 stabilize the anionic group present in most NSAIDs. NSAID aromatic rings are accommodated in the hydrophobic channel. Ser530 is the residue acetylated by aspirin. Note the presence of the relatively bulky Ile523 In COX2 residues Arg120, Tyr355 & Ser530 are present. However, residue 6 is Val523 which allows copening of a side pocket. This pocket accommodates the sulfonamide or isoster of COX2 inhibitors. They are stabilized by hydrogen bonding with Arg513. from Nature Reviews Drug Discovery, 2, 2003 COX-1 & COX-2 - II Overall structure and catalytic activity of both are similar Vivid distinctions in their regulation and expression COX-1 is constitutive and its expression is regulated by hormonal signals involved in maintaining physiologic homeostasis COX-1 is expressed in all tissues Importantly, COX-1 but not COX-2 is constitutively expressed in the stomach, where it is involved in mucosal defense and repair COX-2 expression and activity is largely responsive to adverse stimuli, such as inflammation and physiologic imbalances Control of COX-2 transcription and translation is thought to be the primary mechanism by which steroids such as hydrocortisone and dexamethasone modulate this enzyme. COX-2 has a binding site for steroids whereas COX1 does not COX-2 is constitutively expressed notably in the brain and kidney Mechanism of Action Inhibitors of cyclooxygenase reduce the amount of Prostaglandins and thereby reduce the inflammation process primary insult: Unionized at stomach pH that allows passage into gastric mucosal cells. The inside higher pH ionize them which can not pass through lipid barriers and is trapped inside the cell. This alters the permeability of the cell membranes and allows accumulation of hydrogen ions which cause cell damage Microenvironment low pH AR—COO– + H + AR—COOH Unionized fract ion predom inates Int racellular higher pH AR—COOH AR—COO– + H + Ionized fract ion predom inates The secondary insult is the result of the mechanism of action. The inhibition of PG synthesis prevents the cytoprotective action of the PG Most of the gastric effects of NSAIDS are attributed to their acidic character which participates in 1) decreasing surface hydrophobicity of the mucus gel layer with subsequent loss of barrier properties; 2) uncoupling of oxidative phosphorylation with subsequent increase in mucosal permeability and back diffusion of hydronium ion; 3) ion trapping into the mucosal epithelium Classes of COX Inhibitors The COX inhibitors can be grouped into four classes based on their mechanism of action. 1. Irreversible inhibitors. Aspirin is the only known member of this group 2. Reversible competitive inhibitors of both COX which is freely reversible 3. Slow time dependent inhibition of both COX—they bind and induce a conformational change in the enzyme thus binding very tightly and dissociated very slowly. It can take several seconds to minutes to reach equilibrium between the reversible and pseudo irreversible complex. However, in vivo both mechanism 2 and 3 are essentially the same. 4. Selective reversible competitive inhibitors COX–2. These agents induce a slow conformational change in COX-2 but not in COX-1. The change increases the inhibitor affinity by >10 fold by binding very tightly and dissociating very slowly. Thus the isozyme selective induction of a conformation change in the enzyme leads to potent inhibition of COX-2 that is not seen for COX-1 With the exception of Aspirin, all the NSAIDS are reversible competitive inhibitors Aspirin is a nonreversible inhibitor, for it acetylates the active site which is the basis for its prophylactic use to prevent heart attacks Research suggests a role for PG in CNS transmission and raises the possibility that selective COX–2 inhibitors may modulate CNS function. This is relevant for those COX–2 inhibitors that lack an acidic group and thus can easily pass the BBB. COX–1 provides a cytoprotective role in the stomach and kidneys. It helps maintain the integrity of the mucosal epithelium and inhibition leads to gastric damage, hemorrhage, and ulceration The cytoprotective role in the stomach and kidney is largely due to the vasodilating properties of PGs which enhance mucosal blood flow Thus COX–1 produces prostaglandins that exert cytoprotective roles whereas COX–2 produces prostaglandins involved in inflammation, fever and pain; and COX–2 activation leads to inflammation Thus COX–2 inhibition produces therapeutic effects and COX–1 inhibition produces unwanted side effects. Unfortunately, most NSAIDS are more effective at inhibiting COX–1 than COX– 2 NSAIDs & COX The "classical" nonselective NSAIDs bind to both COX-1 and COX-2, interacting with the hydrophobic channel of the COX isoenzymes Aspirin, unlike other NSAIDs, irreversibly acetylates a serine residue in both COX-1 and COX-2 preventing arachidonic acid from reaching the catalytic site Other nonselective NSAIDs compete directly with arachidonic acid, inhibiting cyclooxygenase activity in a reversible manner Coxibs, the COX-2-selective inhibitors, preferentially bind to and inhibit COX-2. Coxibs are selective agents because they bind COX-1 poorly and in a rapidly reversible manner, whereas they bind COX-2 more tightly Preferential inhibition of COX-2 is thought to be due to the additional space in the COX-2 hydrophobic channel, as well as to the presence of a side pocket in the channel. This side pocket can discriminate the coxibs from nonselective agents based on the different overall structures of these agents, in particular, by the presence in coxibs of specific side chains NSAIDs do not affect the peroxidase site The COX Binding Site 1. A cationic center and two hydrophobic areas 2. The cationic site is attributed to a guanidinium group on Arginine 3. The first hydrophobic area is located adjacent to the cationic center 4. The second region lies under and out of the plane with the first hydrophobic area and is commonly referred to as a trough Some agents can bind only the cationic center and the first hydrophobic area Others can bind all three, resulting in better binding The only way to bind both hydrophobic regions simultaneously is if the drug contains two aromatic ring systems that are perpendicular and not coplanar Binding to the trough can enhance potency. If the ring cannot fit into the trough then it bangs into the walls of the enzyme, sterically inhibiting binding If the two rings are separated by one or more sigma bonds, the two rings may assume a large number of possible conformations due to free rotation around a sigma bond, only a few compliment the receptor. Making rigid molecule with correct conformation gives potent drugs SAR Summary for COX Inhibitors 1. Molecule must have an ionizable acid group and an aromatic ring system 2. A second non coplanar aromatic ring increases potency by increasing bonding interactions 3. Limiting the number of possible conformers increase potency 4. A two atom separation between the anionic charge and the aromatic ring is the optimal 5. Increasing the distance to 3 or 4 carbons generally decreases potency 6. Introduction of a methyl at the first carbon increases potency and introduces a chiral center 7. The S–isomers are the more potent isomers 8. Increasing the size of the alkyl decreases potency but incorporation of the alkyl into a heterocycle retains activity The Salicylates Salicylic acid is a natural product, present in the bark of willow and poplar trees The active ingredient, isolated by a French pharmacist in 1827, was Salicin, oxidized to Salicylic acid OH OH O HO HO O OH Sal ici n In 1875 a Swizz pharmacist, Lowig, distilled meadowsweet flowers and got salicylaldehyde Salicylate SARs The simplest active compound is the salicylic acid anion, The carboxylic group is necessary for activity and the hydroxyl group must be ortho to it. Introduction of electronegative groups and lipophilic groups increases anti–inflammatory activity and toxicity. ASPIRIN SODIUM SALICYLATE (Bisalate) O O OH SODIUM THIOSALICYLATE CHOLINE SALICYLATE Rexolate Anthropan O H3 C O H3C ONa O O ONa OH N CH3 OH SH OH H3C O MAGNESIUM SALICYLATE Magan, Mobidin, (Trisalate) O SALSALATE Salf lex, Disalcid O O O Benorylate OH C O N H O O CH3 O O Mg O O H H OH O O H3C O Salicylic acid and Sodium salicylate were the original products used but required doses which had much gastric irritation and ulceration. Salicylic acid in the unionized form has a bad taste, thus the sodium salt is used more frequently Salsalate and Benorylate are prodrug esters. The sodium salt is freely soluble in water and helps in its dissolution and faster absorption. Salsalate is only half as potent as an analgesic/antipyretic as Aspirin but produces less GI irritation. Salsalate is a diester of salicylic acid and benorylate is esterified with Acetaminophen Salsalate is insoluble in gastric pH but soluble in the small intestines, thus causing less gastric problems. Further, it is useful in hypersensitivity to Aspirin. Hypersensitivity to ASA is a result of acetylated plasma proteins. Since it produces Salicylic acid it can be used in Aspirin sensitive patients Sodium thiosalicylate is used in rheumatic fever and acute gout and an injectable form is available Magnesium salicylate form stable aqueous solution and show some success in overcoming the GI problems Choline salicylate is absorbed faster than Aspirin producing higher salicylate blood levels and an aqueous formulation is available Aspirin Searching for a less toxic better tolerated derivative of salicylic acid produced aspirin. The knowledge that acetylation of the very toxic aniline produced the less toxic acetanilide, acetylation of salicylic acid with acetic anhydride produced Aspirin The name. Aspirin was coined by adding an a for acetyl to spirin from the name of the plant from which salicylic acid was first isolated It is slightly soluble in water, absorbed as such, but is hydrolyzed rapidly to salicylate and acetate by esterases Pharmacological actions are attributed to both the ASA and salicylic acid ASA irreversibly inhibits the enzyme acetylating a serine residue thus preventing access to the cyclooxygenase site Salicylic acid forms a reversible ionic bond with the cationic site on cyclooxygenase NHCOCH2NCO NHCOCH2NCO CH2 O COOH O H CH3 O NHCOCH2NCO CH2 O + H+ COOH O CH3 O- CH2 COOH O OH + O CH3 Salicylamide and Diflunisal SALICYLAMIDE (Bisalate) DIFLUNISAL Dolobid F O O NH2 OH OH F OH Salicylamide is an isostere of salicylic acid, OH replaced by NH2 to produce a non acidic amide which is stable in aqueous preparations and does not cause GI tract ulceration and is absorbed only in intestine. It has greater CNS penetration. It is reported to be as effective as Aspirin as an analgesic/antipyretic and is effective in relieving arthritis pain but does not appear to have antiinflammaatory actions. It does not satisfy SAR 1 possibly works through a different mechanism. It can be used by those allergic to Aspirin. Diflunisal has changed absorption profile and increased duration of action. Diflunisal is absorbed only in intestine; it is not soluble in gastric fluid. Thus, gastric bleeding and GI upset is not as common. It lasts 3–4 times longer than aspirin. The increase in potency is attributed to an increase in binding to the receptor since it has a second aromatic ring SAR 2. The proximity of the two phenyl rings allows for the ortho hydrogen van der Walls electron radii to repel and thus keep the rings out of the same plane. Fenemates The Fenemates are derivatives of Anthranilic acid, an isoster of salicylic acid The most potent analogs are those disubstituted at 2’ and 3’. This indicates that activity resides in compounds with the substituent on the second ring that keep it out of coplanarity by the ortho substituent Mefenamic acid has only one substituent, the 2’ methyl, that ensures non coplanarity Meclofenamate sodium has two such groups, the chlorine atoms, and thus more molecules of Meclofenamate assume the correct conformation and the drug is more potent MEFENAMIC ACID Ponstel O MECLOFENAMATE Sodium Meclomen O OH ONa NH NH CH3 CH3 Cl Cl CH3 Meclofenamate is 25 times more potent thus normal dose for Meclofenamate is 25 mg while the dose for Mefenamic acid is 250 mg. Since this class offers no advantage over the salicylates with respect to analgesic or anti-inflammatory actions, there is little interest in developing this class p-Aminophenols O H N O CH3 H N O CH3 Phenacetin Acetanilide O CH3 H N CH3 ACETAMINOPHEN Tylenol, Datril, Panadol OH Liquiprin, Tempra Useful for pain and fever, but not inflammation. They have an aromatic ring, but do not have an acidic group ionizable at physiologic pH. Thus they do not comply with SAR 1 possibly act by some other mechanism The first drug Acetanilide is out of market due of toxicity (both blood and liver disorders Phenacetin (1887) was used for decades, but in the 1970s it was implicated in cases of liver and nephrotoxicity and was removed from the market Acetaminophen is also a very old drug, a metabolite of both phenacetin and acetanilide, is a safe drug, producing much better tolerance and a lower incidence of gastric bleeding compared to many of the other NSAIDs, probably because of its apparently different mechanism of action You know the chemistry of toxicity for both phenacetin and acetaminophen Pyrazoles and Pyrazolidinediones ANTIPYRINE (Auralgan Otic) Antipyrine is the prototype and its antipyretic and analgesic activities were discovered by accident. Aminopyrine O O CH3 N N N N CH3 H3C N H3C CH3 CH3 Aminopyrine is an analog, more potent and longer acting but both possess significant incidences of agranulocytosis leading to death and used only in otic drops Dipyrone is a prodrug which spontaneously decomposes in aqueous solutions to aminopyrine. It is banned in the US but available in Mexico. S ONa N H3C Dichloralphenazone is a complex of Aminopyrine and Chloral hydrate It is a common agent in many OTC analgesics. It is a mild sedative used in migraine /tension headache products. O O N N O CH3 CH3 Dipyrone O N Cl N Cl • Cl OH H3C Although it appears that SAR 1 does not apply, these OH CH3 drugs are able to tautomerize into enols, which in turn DICHLORALPHENAZONE ionize. Thus they have an aromatic ring with an anionic Antipyrine • Chloral Hydrate charge two atoms away. O Search for better drug produced the pyrazolidinediones which are acidic because the bdiketone tautomrizes into an acidic enol N NH pyrazole HN NH pyrazolidine O O OH HN NH HN NH pyrazolidinedione O O N N CH3 N O O Phenylbutazone CH3 N HO Oxyphenbutazone Phenylbutazone is equipotent to antipyrine, and more potent than aspirin for treating inflammation. It has long half-life (72-84 hours). Serious toxicities, e.g., agranuylocytosis, peptic ulcers and bone marrow depression, limits its use in long term therapy. Oxyphenbutazone is its active metabolite with similar activity, equipotent but less toxic, shorter half–life (half-life 48-72 hours) and better tolerated. w-1 Hydroxyl is another metabolite with uricosuric activity but little anti– inflammatory activity The keto metabolite, Kebuzone, is marketed in Europe as a uricosuric agent. Sulfinpyrazone is marketed in the US as a uricosuric. The two phenyl rings are not coplanar due to their close proximity, on adjacent nitrogens. The ortho hydrogens one each ring are effective at this close proximity Arylacetic acid Derivatives Satisfy SAR 1, SAR 2, SAR 4 as well as SAR 3 thus are generally more potent than ASA. Indomethacin was synthesized in 1961 at Merck as part of a study of indole derivatives as potential anti–inflammatory agents since Serotonin, which contains the indole nucleus, is a potential mediator of inflammation. The indole system and the phenyl ring are separated by one atom and thus two sigma bonds. Theoretically it could exist in millions of conformation, but it does not due to skillful molecular manipulations. CH3 O INDOMETHACIN Indocin O H3C O C CH3 O Cl CH3 O CH2 COOH CH3 N OH N CH2 COOH O N O CH3 C Different conformers of Indomethacin Cl Cl Illustrates SAR 3. Partial double bond character of amide restrict rotation. 2-Methyl provides steric hindrance favoring the active conformer and the hydrogen atoms at 7 and 2’ provide hindrance to ensure non coplanarity Sulindac: Indomethacin has significant CNS side effects due to the indole nucleus. Thus the heterocyclic nitrogen was removed and a double bond introduced, giving the indene derivative. Z isomer is active, lacks the CNS side effects and causes less GI irritation but low water solubility. Introduction of a fluoro and a methylsulfinyl increased solubility while retaining potency. Sulindac is a prodrug. Its active form is the sulfide metabolite which has a long half–life allowing for BID administration. The phenyl is out of the plane CH3 O F CH2 COOH CH3 C H Cl Indene analog of Indomethacin OH CH3 H H3 C O CH3 C H CH3 S Sulindac Clinoril CH2 COOH CH3 C S O F CH2 COOH O F H CH3 S Sul indac Sul fide of Sul indac Clinically it has only about half the potency of Indomethacin in treating inflammation and reducing fever, but is equipotent in analgesic effect. Since the drug is absorbed as the inactive sulfoxide, it causes fewer GI disturbances (no prostaglandin biosynthesis inhibition in the stomach). The thioether metabolite (shown at the right) is longer-lived than the parent (ca. 16 hours). Indole replaced with pyrrole N O O ONa ONa CH3 CH3 O H3C TOLMETIN Na Tolectin N CH3 O Cl Zomepirac Na [Zomax] Tolmetin was designed to contain the three portions of Indomethacin deemed necessary for activity, the carboxyl, the flat indole ring and an out of plane phenyl Compared to Indomethocin, is there anything to ensure SAR 3, that one conformer predominates and the two aromatic rings are non coplanar? Which is more potent and why? Tolmetin’s major metabolite is the carboxylic acid resulting from benzylic hydroxylation and subsequent oxidation. It has a half–life of 30 to 60 minutes To increase the duration of action the methyl was replaced with a chloro which prevented metabolism at the phenyl ring. This drug was Zomepirac which was marketed but eventually removed due to reports of severe anaphylactoid reactions in patients sensitive to Aspirin. O O OK ONa NH NH Cl Cl Cl Cl Diclof enac Na Diclof enac K Voltaren, (Arthrotec), Cataf lam Solaraze The SARs in Diclofenac sodium are similar to those discussed with the Fenemates. Diclofenac is probably the most popular NSAID in the world. Its mechanism of action may be a little different from the others. It is a COX inhibitor like the rest, but it also seems to inhibit lipoxygenase to some degree. This could account for its increased anti-inflammatory effectiveness and potency. The two Cl groups are necessary to force the two rings out of plane with each other. It has a profile of action similar to the others and favors anti-inflammation uses, rather than analgesic uses. Diclofenac is also available in combination with Misoprostol as Arthrotec™. Why? The Sodium salt is a delayed release formulation while the Potassium salt is used in a rapid release formulation. Diclofenac sodium is available in a gel form (Solaraze) for the treatment of actinic keratosis. The mechanism is unknown. Arylpropioanic acid Derivatives These agents illustrate SAR 4, 6 and 7 Activity resides in the S isomer. in vivo some of the inactive R isomer is converted to the active S by isomerases, but not the S to R. One reference states that 60% of an Ibuprofen and 100% of a Fenoprofen dose undergo isomerization. Another reference states that S–Ibuprofen is 160 times more active than R–Ibuprofen in vitro but they were equipotent in vivo. IBUPROFEN Motrin, Rufen, Advil, Nuprin, CH3 (Vicoprofen) 3 O O O OH OH OH F H3C KETOPROFEN Orudis, Oruvail CH3 FLURBIPROFEN Ansaid CH O CH3 Ibuprofen is the prototype, marketed as the racemate. Lacks second aromatic ring (SAR 2) but possess a sec–butyl substituent that presumably renders the drug slightly less potent. Its profile is much like other NSAIDs in terms of GI distress. Flurbiprofen resulted from a study of the SARs. The 3–fluoro substituent helps ensure non–coplanarity. This compound had the most favorable therapeutic profile and was first introduced as a topical product for ophthalmic use (Ocufen). Later it was introduced for systemic use (Ansaid is reputed to stand for Another NSAID). This drug is many times the potency of the other drugs (100x phenylbutazone against inflammation), and is about half as potent as methylprednisolone (an anti-inflammatory steroid). Ketoprofen (1986): The great potential advantage of this drug is that it inhibits the leukotriene pathway as well, although its structure does not predict that. It is clinically less potent than Indomethacin, but has about the same GI disturbance profile SUPROFEN Profenal FENOPROFEN Calcium Nalfon CH3 CH3 O O OH S O KETOROLAC Tromethamine Toradol, Acular Ca ++ O O N O NH3 O O HO OH OH 2 Suprofen (1985) is an isostere of Ketoprofen an analgesic for mild to moderate pain. It was found to cause flank pain and transient renal failure and was withdrawn in 1987. It then was reintroduced in 1989 for ophthalmic use in lens replacement surgery to prevent iris inflammation. Fenoprofen (1976) is less potent than many of the others for inflammation, with some analgesic and antipyretic activity. It does not illustrate SAR 3. No special advantage is shown by this drug. Ketorolac is related to Indomethacin and Tolmetin because it has the pyrrole ring, but is a cyclic propionic acid derivative (SAR 8), commercially available as the tromethanime salt. The tromethamine moiety enhances water solubility. The injectable formulation is incompatible with solutions of conjugate acids like meperidine hydrochloride (precipitation). It is about half as potent as Morphine when injected. After its success as a parenteral agent, an oral agent was marketed. NAPROXEN Naprosyn NAPROXEN Sodium Anaprox, Aleve, CH3 Naprelan CH3 O O CH3 OH O CH3 CARPROFEN Rimadyl O CH3 O ONa Cl OXAPROZIN Daypro OH O O NH N OH Naproxen does not possess a second non coplanar ring (SAR 3). The naphthyl rings are fused and aromatic thus flat and planar. It is the only drug currently marketed in the optically pure form. This is not due to resolution but is the result of the synthetic method used. Interestingly the S isomer of Naproxen is (+) as most in this class are, but the S isomer of the sodium salt is (–). Carprofen is marketed as a veterinary analgesic. Does it have a second non–coplanar ring, SAR 2? Oxaprozin is an aryl propionic acid but is unique in that the propyl is not branched. Oxicams Pfizer developed this class to produce non–carboxylic acid NSAIDS PIROXICAM Feldene OH N O S O MELOXICAM Mobic OH O O N H CH3 N N O S N N H S CH3 CH3 O Piroxicam is the first member of this family marketed, however it possess the three structural requirements. The enolic hydroxyl is the acidic group and the pyridyl ring is the second aromatic ring. Although it has good potency, the GI side effects limit its usefulness. A typical half-life for Piroxicam is ca. 38 hours. Meloxicam is structurally related to Piroxicam. Although Meloxicam is frequently described in the literature as a selective COX-2 inhibitor, it is considerably less selective for the COX-2 versus COX-1 isoenzyme when compared to Celecoxib or Rofecoxib. Miscellaneous ETODOLAC Lodine H3C H3C H N NAMBUMETONE Relafen O 6–Methoxy–2–naphthylic acid 6–MNA O O O OH O O CH3 CH3 OH Etodolac can be considered a nonclassical bioisostere of the arylpropionic acids. It is ca. 50x more potent than aspirin in inflammation, 33% as potent as indomethacin. It shows a much better GI profile than aspirin or indomethacin and this can be a therapeutic advantage. It is a unique compound. How many aromatic rings does it have? Note the separation between the aromatic ring and the acid. Nabumetone is a ketone and thus non–acidic (SAR1?). It is classed as an Alkanone. It is a prodrug and must be activated by b–oxidation to 6–Methoxy–2– naphthylacetic acid. Approximately 35% of a 1000mg dose is converted to 6– MNA, which is structurally related to Naproxen, an arylacetic acid. But is only an acetic acid derivative thus weaker than Naproxen. Further, not all the dose is converted to 6–MNA. The advantages of this drug is less GI tract toxicity because it is not acidic Allopurinol is a structural analog of hypoxanthine and thus is a xanthin oxidase inhibitor used to treat hyperuricemia and its complications including chronic gout as well as prophylaxis with chemotherapeutic treatments, which can rapidly produce severe hyperuricemia. O O N HN HN N N N H Hypoxanthene N N H Allopurinol Drugs for Inflammatory Bowel Disease (Ulcerative Colitis) Mesalamine or 5-aminosalicylic acid (5-ASA), and its prodrugs balsalazine and olsalazine are anti-inflammatory drugs/prodrugs used to treat inflammation of the digestive tract (ulcerative colitis) and mild-to-moderate Crohn's disease. Mesalazine is a bowel-specific aminosalicylate drug that acts locally in the gut and has its predominant actions there, thereby having few systemic side effects. Balsalazine and olsalazine generate mesalamine in the site of action. (How about salfasalazine??) NaOOC H2N NaOOC O OH HO N N OH HO N N Mesalamine COOH HN Olsalazine COONa O Balsalazine ONa COX - 2 Inhibitors CELECOXIB Celebrex F 3C O N N S NH2 O O REFECOXIB Vioxx VALDECOXIB O Bextra O S O CH3 CH3 O N S NH2 DERACOXIB Deramax O S F N O F O CH3 NH2 O N F OCH3 Celecoxib was the first. Structurally it differs from other NSAIDS in that is only weakly acidic. It does possess a sulfamyl group and has a warning about use in patients with a sulfonamide allergy Valecoxib is also a sulfamyl and its package insert contains the same caution. It is this phenyl group which is inserted into the extra space in COX–2 Deracoxib is also acidic but is indicated for veterinary use Rofecoxib is not acidic Refecoxib and MI One of the reasons given is that the endothelial cells express mainly COX–2 whereas platelets express COX–1. Since Refecoxib is COX– 2 selective it allows for an overproduction of Thromboxane A2 (platelet). Thromboxane is released by platelets and causes vasoconstriction and platelet aggregation. Prostacycline is release by capillary endothelium and causes vasodilatation and prevents platelet aggregation. Normally these balance each other; The platelets use COX-1 and the capillaries use COX-2. Thus COX-2 selective agents unbalance the system favoring thromboxane. However this is not the only factor in play since the other COX–2 selective agents have not shown the increase in MI Study Guide What are cyclooxygenase and peroxydase? What do you mean by prostaglandin endoperoxide H2 synthase (PGHS)? What are COX-1 and COX-2? What physiologically important prostaglandins and thromboxanes are synthesized by them blocking of which gives clinical effects of different NSAIDs? What are different chemical categories of NSAIDs? List the nonselective and COX-2 selective NSAIDs with structures. Similarities and differences between COX-1 and COX-2 – with reference to physiology, active site amino acids, size and shape of active site. Diflunisal is a salicylic acid derivative yet does not cause much is gastric bleeding and GI upset. Why? Why low-dose, long term aspirin is recommended for the prevention of strokes and heart attacks? Describe the action and mechanism of action of acetaminophen. Is COX-3 important for its activity? Study Guide Cont. Are pyrazoles & pyrazolidinediones acids? What is their mechanism of action? Why indomethacin is highly potent analgesic yet toxic to CNS? Why sulindac can be considered as its isostere without CNS effect? Is sulindac a prodrug? Why diclofenac and ketoprofen are highly potent anti-inflammatory agents? Which stereoisomer of ibuprofen is biologically active? Why it is not necessary to resolve the active form rather than administering the recemic mixture? The mercapturic acid conjugate is a sign of acetaminophen toxicity. Why? Are the oxicams classified as COX-2 selective agents? Why or why not? What is the active principle of sulfasalazine in IBS? Why it is replaced with better alternatives? Which are they? Why coxibs are selective to COX-2 and not bound to COX-1. Why the coxibs have more CV risk than other NSAIDs?