MS PowerPoint
advertisement

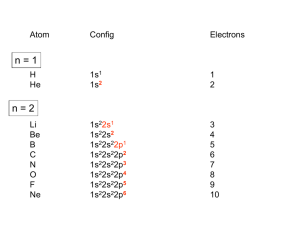
HOMOGENEOUS CATALYSIS CHOICE OF METAL LIGANDS AND BASICS A.V. Ramaswamy Email: av_ram@hotmail.com HETEROGENEOUS 1. All Catalytic processes 2. TON/TOF 3. Selectivity (Chemo-, Regio-, and Enantio-) 4. Kinetics 5. Mechanistic Understanding 4. Thermal Stability 5. Catalyst Life 6. Catalyst recovery/Separation 7. Heat recovery 8. Susceptibility to poisons 9. Macroscopic Diffusion 10. Applications 11. Catalysts/Costs 85% Similar High with zeolites for Regio-selectivity Could be complex Surface reaction Not simple, -Interface -surface structure - Non-homogeneity High Temperatures Longer High Design More Important - Petroleum refining - Petrochemicals, bulk Chemicals and intermediates High volumes, lower costs HOMOGENEOUS 15% Very High for Enantio-selectivity Somewhat simpler Performance can be easily explained and understood at molecular level < 250oC Shorter Difficult/Serious problems Simpler Less susceptible Not important - Fine Chemicals - Pharmaceutical intermediates Tailor-made plastics Lower volumes and more expensive A Historical Perspective Oldest Metallo enzymes Yeast whole cell Fe porphyrin Zn complexes Ni complexes in Hydrogenase enzyme Cobalt corrin complexes Man-made 1750 Lead chamber process 1920 Mercury sulfate 1950 Co complexes Ni (DuPont) Co (BASF) (Shell) Mo 1970 Rh (Monsanto) (Wilkinson) whether they are truly homogeneous? fermentation of sugars Oxidation decabrboxylation H2 activation C-C bond formation SO2 to SO3 by N oxides (Homogeneous ?) acetylene to acetaldehye (Wacker process) oligomerization of ethene hydrocyanation Carbonylation Hydroformhylation Epoxidation of propene Carbonylation Hydrogenation Lighter olefins Asymmetric hydrogenation to L-Dopa Ring opening polymerization Homogeneous Catalysis – Recent, narrower definition Involves (organo)-metallic complexes as the catalysts, should contain a bond between a carbon atom and the metal The reactions employing homogeneous catalysts that are not (organo) metallic Complexes: -General acid-base reactions (ester hydrolysis) -Lewis acids as catalysts (Diels-Alder reaction) -Organic catalysts (thiazolium ions in Cannizaro reaction) -Porphyrin complexes (epoxidation, hydroxylation) -Enzymatic processes -Coordination complexes (polyester condensation) Ligand Effects are extremely important in homogeneous catalysis by metal complexes Example: Various products from 1,3-butadiene with a “Ni” catalyst, where by changing the type and the nature of the ligands coordinated to “Ni” , dimerization, oligomerization and polymerization reactions take place Effect of ligands and valance states on the selectivity in the nickel catalyzed reaction of butadiene ( ( ) ) n ( )n n Scheme: 1,3-butadiene reactions on “Ni” Ligand Effects A. Electronic Effects P as donor element: Alkyl (aryl) phosphines and organo phosphites Alkyl phosphines are strong bases, good σ-donor ligands Organo phosphites are strong π-acceptors and form stable complexes with Electron rich transition metals. Metal to P bonding resembles, metal to ethylene and metal to CO Which orbitals of P are responsible for π back donation? Antibonding σ* orbitals of P to carbon (phosphine) or to oxygen (phosphites) The σ-basicity and π-acidity can be studied by looking at the stretching frequency of the coordinated CO ligands in complexes, such as Ni L(CO)3 or Cr L(CO)5 in which L is the P ligand. 1) Strong σ donor ligands → High electron density on the metal and hence a substantial back donation to the CO ligands → Lower IR frequencies Strong back donation and low C – O stretch 2) Strong π acceptor ligands will compete with CO for the electron back donation and C-O stretch frequency will remain high Weak back donation → High C – O stretch The IR frequencies represent a reliable yardstick for the electronic properties of a series of P ligands toward a particular metal, M. CrL(CO)5 or NiL(CO)3 as examples; L = P(t-Bu)3 as reference The electronic parameter, χ (chi) for other ligands is simply defined as the difference in the IR frequencies of the symmetric stretch of the two complexes Ligand, PR3, R= χ (chi) IR Freq (A1) of NiL(CO)3 in cm-1 T-Bu N-Bu 4-C6H4NMe3 Ph 4-C6H4F 0 4 5 13 16 2056 2060 2061 2069 2072 CH3O PhO CF3CH2O Cl (CF3)2CHO F CF3 20 29 39 41 54 55 59 2076 2085 2095 2097 2110 2111 2115 B. Steric Effects 1) Cone angle (Tolman’s parameter, θ) (Monodentate ligands) From the metal center, located at a distance of 2.28 A from the phosphorus atom in the appropriate direction, a cone is constructed with embraces all the atoms of the substituents on the P atom, even though ligands never form a perfect cone. Sterically, more bulky ligands give less stable complexes Cone angle Crystal structure determination, angles smaller than θ M P values would suggest. Thermochemistry: heat of formation of metal-phosphine adducts. When electronic effects are small, the heats measured are a measure of the steric hindrance in the complexes. Heats of formation decrease with increasing steric bulk of the ligand. Ligand, PR3; R = H CH3O n-Bu PhO Ph i-Pr C6H11 t-Bu θ value = 87 107 132 128 145 160 170 182 An ideal separation between Steric and electronic parameters is not possible. Changing the angle will also change the electronic properties of the phosphine ligand. Both the - and θ- values should be used with some reservation Predicting the properties of metal complexes and catalysts: Quantitative use of steric and electronic parameters (QALE) The use of - valaues in a quantitative manner in linear free energy relationships (LFER) Tolman’s equation: Property = a + b() + cθ The property could be log of rate constant, equilibrium constant, etc. Refinements: Property = a + b () + c(θ – θth) Where, , the switching factor, reads 0 below the threshold and 1 above it. Refinement, the electronic parameter: Property = a(d) + b(θ – θth) + c(Ear) + d(p) + e Where d is used for -donicity and p used for -acceptor property; Ear is for “aryl effect”. For reactions having a simple rate equation, the evaluation of ligand effects with the use of methods such as QALE will augment our insight in ligand effects, a better comparison of related reactions, and a useful comparison between different metals. Bite angle effects (bidentate ligands) Diphosphine ligands offer more control over regio- and stereoselectivity in many catalytic reactions The major dfiference between the mono- and bidentate ligands is the ligand backbone, a scaffold which keeps the two P donor atoms at a specific distance. This distance is ligand specific and it is an important characteristic, together with the flexibility of the backbone P O P P P X X P P P P X Many examples show that the ligand bite angle is related to catalytic performance in a number of reactions. Pt-diphosphine catalysed hydroformylation Pd catalyzed cross coupling reactions of Grignard reagents with organic halides Rh catalyzed hydroformylation Nickel catalyzed hydrocyanation and Diels-Alder reactions Kinetic studies Reaction rates: Dependence on the concentration of reactants (and the products in some cases) Useful in understanding the mechanism of the reaction Empirically derived rate expressions Ligand dissociation: leads to generation of catalytic active intermediate. If the ligand is added to such a catalytic system, the rate of the reaction decreases. Examples: CO dissociation in Co-catalysed hydroformylation Phosphine dissociation in RhCl(PPh3) catalysed hydrogenation Cl- dissociation in the Wacker process Michaelis-Menten Kinetics (Enzyme catalysed reactions) Saturation kinetics Rate = k.K[substrate][catalyst]/1 + K[substrate] A complex is formed between the substrate and the catalyst by a rapid equilibrium reaction. The equilibrium constant of this reaction is K, and it is followed by the rate-determining step with a rate constant, k. Increasing the substrate concentration will increase the rate initially, followed by more or less constant rate high substrate concentration, when K[substrate] ~ 1 + K[substrate] at constant catalyst concentration, a plot of (1/rate) vs. (1/(substrate) will give a straight line. Creation of ‘vacant site’ and coordination of the substrate The function of a catalyst is to bring the reactants together and lower the activation barrier for the reaction. To bring the reactants together, a metal center must have a vacant site. Compare a homogeneous catalyst and a solid heterogeneous catalyst. The latter has a surface. Homogeneous catalyst in a condensed phase: How do we create a vacant site? The solvent molecules will always be coordinated to the metal ion. There is a competition between the desired substrate and the other potential ligands present in the solution. Classical way to look: Substitution reactions Associative and Dissociative mechanisms Basic Chemical Concepts A metal complex: The catalytic activity is influenced by the characteristics of the central metal ions and attached ligands. The metal: The oxidation state and the electron count of the valence shell of the metal ion. A fully ionic model is implicit. Oxidation state PPh3 Cl Rh Ph3 P 1+ Electron count 16 1+ 18 4+ 16 1- 18 PPh3 H Ph3 P PPh3 Rh PPh3 CO + CH3 Zr O CO OC Co OC CO Rule of effective atomic number (EAN) or the 18 e- rule Coordinative unsaturation High reactivity Reasons When the electron count is less than 18, the metal complex is often classified as Coordinatively unsaturated. Easy displacement of weakly bound ligands; e.g., Zr Complex, THF can be easily replaced by the substrate and solvent molecules. Influenced by bulkier ligands; Steric constraints NiL4 ↔ NiL3 + L Many complexes have electron counts less that 16 Important properties of ligands 1. Ligands: CO, R2C=CR1, PR3 and H- (N2, NO, etc.) All ligands behave as Lewis bases and the M acts as a Lewis acid Alkenes: electrons Whereas H2O and NH3 accept e- density from the metal, i.e., they act as Lewis Acids ( acid ligands) The donation of e- density by the metal atom to the ligand is referred to as back donation. H2 acts as a Lewis acid. Also, Lewis acid-like behaviour of CO, C2H4 and H2 in terms of overlaps between empty orbitals of the ligand and the filled metal orbitals of compatible symmetry. Back donation is a bonding interaction between the metal atom and the ligands, because the signs of the donating metal ‘d’ orbitals and the ligand * (* for H2) acceptor orbitals match. The ligands play important roles in a large number of homogeneous catalytic reactions. 2. Another set of ligands: Alkyl, Allyl and alkylidene ligands Alkyl ligands: Two reactions a) Addition of RX to unsaturated metal center M + R R M X Oxidation state: +n valence electrons: p X +n+2 p-2 b) Insertion of alkene into a metal-H or an existing metal-C bond M R R H H M H H Reactivity of metal-alkyls: kinetic instability towards conversion by -hydride elimination. Others: H H H -hydride elimination R M Agostic interaction Metallocycle formation M R H M + R X H R M M R R M H H H H X H H H M R M R H H OR M M R R H M R M R M H M Important Reaction Steps Homogeneous Catalysis There are 5 types of reactions (and their reverse) which, in combination, account for most homogeneous catalytic cycles involving hydrocarbons 1. Ligand Coordination and Dissociation 2. Insertion and Elimination 3. Nucleophilic attack on coordinated ligands 4. Oxidative addition and Reductive elimination 5. Oxidation and Reduction Important Reaction types 1. Oxidative addition and Reductive elimination Electronic ligand effects are highly predictable in oxidative addition reactions; -donors (alkylphosphines) strongly promote the formation of high-valence states and thus oxidative additions. Compexation of halides to Pd(0) increases e- density and facilitates oxidative addition. Phosphites and CO, on the other hand, reduce e- density on the metal and thus the oxidative addition is slower or may not occur at all. Reductive elimination is simply the reverse reaction of oxidative addition. The formal valence state of the metal is reduced by two. And the total electron count of the complex is reduced by two. In a catalytic cycle, the two reactions always occur pair-wise. Reductive eliminations can be promoted by stabilization of the low-valent state of the product. This means ligands that are good -acceptors, bulky ligands, and ligands preferring bite angles more suited for tetrahedral than for square planar complexes, when we deal with group 10 metals. 2. Migratory insertion Reactions In homogeneous catalytic reactions, old bonds are usually broken by Oxidative addition reactions and New bonds are formed by Reductive elimination and insertion reactions. How does insertion take place ? A more appropriate description of a number of insertion reactions is “migratory insertion” 2. Insertion reactions : Migratory insertion - Examples H H M M R M R M CO CO R M Insertion of CO into M-R bond O H M Insertion of olefin into M-R bond O R M Insertion of olefin into M-H bond M H Insertion of CO into M-H bond Insertion reactions are ‘cis’ in character M H M H O R CO M M R 3. -Hydride elimination This is also called -elimination reactions: Hydride abstractions can occur from carbon atoms in other positions also, But elimination from the -carbon is more common. Insertion of an alkene into a metal-hydrogen bond and -elimination reaction have a reversible relationship. It is possible to study this reversible equilibrium by NMR spectroscopy. e.g., a hydrido-ethylene complex of Rh L H L Insertion Rh Rh ß-elimination L L L = PPr3i M M H n H + Polymer chain termination by ß-elimination n 4. Nucleophilic attack on a coordinated ligand Upon coordination to a metal center, the electronic environment of the ligand undergoes a change. The ligand may become susceptible to electrophilic or nucleophilic attack. Pd 2+ + H2O OH [ Pd + ]+ H+ R Ti 4+ O + O Ti 4+ R H H O Fe CO O + O + HO- - Fe OH The extent of the reactivity of the ligand is reflected in the rate constants 5. Oxidation and Reduction During a catalytic cycle, metal atoms frequently alternate between two oxidation states: Cu2+/Cu+ Co3+/Co2+ Mn3+/Mn2+ Pd2+/Pd Catalytic Oxidation: generating alcohols and carboxylic acids The metal atom 1) initiates the formation of the radical R• 2) contributes to the formation of R-O-O• radical R H + Co(III) R R O O H + Co(II) + O2 R + H + Co(II) R O O R O + Co(III)OH R H AND R O O H + R R O O H + Co(III) R O O + H + Co(II) The Catalytic Cycle and the Intermediates Example: A metal complex catalyzed hydrogenation of an alkene → Alkene + H2 Alkane MLn+1 ⇋ MLn + L MLn+ + H2 ⇋ H2MLn H2MLn + alkene ⇋ H2MLn(alkene) H2MLn(alkene) ⇋ HMLn(alkyl) HMLn(alkyl) → MLn + alkane Catalytic Cycle and Catalytic Intermediates a. Kinetic studies and mechanistic insight i) Macroscopic rate law ii) Isotope labelling and its effect on the rate or stoichiometry iii) Rate determining step iv) Variation of ligand structure and its influence on ‘k’ b. Spectroscopic investigations ‘in-situ’ IR, NMR, ESR c. Studies on model compounds d. Theoretical calculations Limitations: - Kinetic studies are informative about the slowest step only, not other steps. - Spectroscopic investigations of a complex requires a minimum concentration. - It is possible that the catalytically active intermediates never attain such concentrations and therefore, not observed. -The species that are seen by spectroscopy may not be involved in the catalytic cycle! However, a combination of kinetic and spectroscopic methods can resolve such uncertainties to a large extent. Reference Books 1. Homogeneous Catalysis: The Applications and Chemistry of Catalysis by soluble Transition Metal Complexes, G.W. Parshall and S.D. Ittel, Wiley, New York, 1992. 2. Applied Homogeneous Catalysis with Organometallic Compounds, Vols 1 & 2, edited by B. Cornils and W.A. Herrmann, VCH, Weinheim, New York, 1996. 3. Homogeneous Catalysis: Mechanisms and Industrial Applications, S. Bhaduri and D. Mukesh, Wiley, New York, 2000. 4. Homogeneous catalysis: Understanding the Art, Piet W.N.M. van Leeuwen, Kluwer Academic Publishers, 2003.