Strong dependency of reaction rates on coordination number of
advertisement

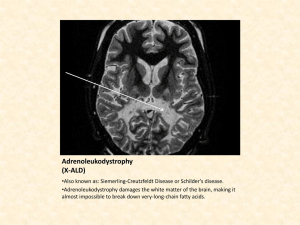
Multi-Scale modelling of atomic layer deposition Presented by: Mahdi Shirazi Supervised by: Dr. Simon Elliott Outline • Atomic layer deposition (ALD) • • • • • Description of ALD Goals Obstacles Findings from atomic-scale modelling Strategy for Kinetic Monte Carlo What is ALD? ALD is based on self-limiting surface reactions of two chemicals. For an oxide, a metal precursor & an oxygen precursor. Process is cyclic : 1. Pulse of metal precursor - a monolayer of metal precursor molecules chemisorbs onto surface . 2. Purge - to remove unreacted precursor and by-products from chamber. 3. Pulse of oxygen precursor – to create a monolayer of chemisorbed oxygen precursor on surface 4. Purge – to remove unreacted precursor and by-products from chamber. www.isr.umd.edu/~hennlec/images/ALD/ALD_reaction_475.jpg The desired film thickness is reached by repeating the cycle. Typical growth per cycle is about 0.1 nm/cycle and cycle time is typically 1-4 s/cycle. Goals • Model interaction of precursors with surface and growth by ALD. • Go beyond the atomic length scale and the time scale of individual reactions. • Explain why amorphous or crystalline layers are deposited? Obstacles • Reaction mechanisms consist of rare events. • Need to evaluate system beyond picosecond timescale. • Using density functional theory (DFT) is time consuming.(e.g. Cl-NEB calculation takes 3 hours for 300 atoms by 120 Intel-Xeon CPU). • To find reaction events: how efficient are Nudged Elastic Band (NEB), Conjugate Gradient (CG), quasi-Newton algorithms and ab initio Molecular Dynamics (MD)? We should take advantage of Kinetic Monte Carlo (KMC) to describe ALD. Outline • Atomic layer deposition • Findings from atomic-scale modelling • • • • ALD reactions Non-ALD reaction Evaluating barriers by NEB Importance of coordination number • Strategy for Kinetic Monte Carlo Slab modelling • • • • • Growth of HfO2 from Hf(N(CH3)2)4 and H2O Monoclinic structure is stable phase in low temperature. Direction of growth (111) Four layers have been regarded as slab Extended surface 22 We used hydroxylated surface VASP code. Slab=yellow, oxygen=red, hydrogen=white • • VASP: http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html • • • H2O dissociates at active Lewis acid and base sites at surface Cover the surface with hydroxyl groups and water molecules Rate of proton diffusion depends on coverage of OH. 1-Charles B. Musgrave et al., Chem. Mater. 2006, 18, 3397-3403. Hafnium =grey, oxygen=red, hydrogen=white Adsorption and dissociation of H2O at HfO2 surface1 Hafnium =blue, oxygen=red, hydrogen=white, Nitrogen= darkblue, carbon=grey Sequence of ALD reactions Barriers were calculated by Cl-NEB1 HfX4+2H2O 4HX+HfO2 X=N(CH3)2 We used DFT2,3 to calculate activation energies to implement them into the KMC 1. Graeme Henkelman, Hannes Jo´nsson et al J. Chem. Phys.113, 22, 9901 2000 2. VASP: http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html 3. Entropy calculated by TURBOMOLE http://www.turbomole.com/ T=500K Hafnium =blue, oxygen=red, hydrogen=white, Nitrogen= darkblue, carbon=grey Sequence of ALD reactions Barriers were calculated by Cl-NEB1 HfX4+2H2O 4HX+HfO2 X=N(CH3)2 We used DFT2,3 to calculate activation energies to implement them into the KMC 1. Graeme Henkelman, Hannes Jo´nsson et al J. Chem. Phys.113, 22, 9901 2000 2. VASP: http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html 3. Entropy calculated by TURBOMOLE http://www.turbomole.com/ T=500K Hafnium =blue, oxygen=red, hydrogen=white, Nitrogen= darkblue, carbon=grey Sequence of ALD reactions Barriers were calculated by Cl-NEB1 HfX4+2H2O 4HX+HfO2 X=N(CH3)2 We used DFT2,3 to calculate activation energies to implement them into the KMC 1. Graeme Henkelman, Hannes Jo´nsson et al J. Chem. Phys.113, 22, 9901 2000 2. VASP: http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html 3. Entropy calculated by TURBOMOLE http://www.turbomole.com/ T=500K Hafnium =blue, oxygen=red, hydrogen=white, Nitrogen= darkblue, carbon=grey Sequence of ALD reactions Barriers were calculated by Cl-NEB1 HfX4+2H2O 4HX+HfO2 X=N(CH3)2 We used DFT2,3 to calculate activation energies to implement them into the KMC 1. Graeme Henkelman, Hannes Jo´nsson et al J. Chem. Phys.113, 22, 9901 2000 2. VASP: http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html 3. Entropy calculated by TURBOMOLE http://www.turbomole.com/ T=500K Hafnium =blue, oxygen=red, hydrogen=white, Nitrogen= darkblue, carbon=grey Sequence of ALD reactions Barriers were calculated by Cl-NEB1 HfX4+2H2O 4HX+HfO2 X=N(CH3)2 We used DFT2,3 to calculate activation energies to implement them into the KMC 1. Graeme Henkelman, Hannes Jo´nsson et al J. Chem. Phys.113, 22, 9901 2000 2. VASP: http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html 3. Entropy calculated by TURBOMOLE http://www.turbomole.com/ T=500K Hafnium =blue, oxygen=red, hydrogen=white, Nitrogen= darkblue, carbon=grey Sequence of ALD reactions Barriers were calculated by Cl-NEB1 HfX4+2H2O 4HX+HfO2 X=N(CH3)2 We used DFT2,3 to calculate activation energies to implement them into the KMC 1. Graeme Henkelman, Hannes Jo´nsson et al J. Chem. Phys.113, 22, 9901 2000 2. VASP: http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html 3. Entropy calculated by TURBOMOLE http://www.turbomole.com/ T=500K Barrier to H+ diffusion to amide group Cl-NEB • Our calculations showed that H+ diffusion barrier varies between 0 to 2eV. 3.71eV Barrier to H+ diffusion to amide group Cl-NEB Test whether amide ligand can desorb without combining with H+? No: barrier increases from 1.6 eV to 3.7 eV in absence of H+. Discovery of reaction events (MD superior to optimisation) Densification1 Ligand transfer show role of coordination number 1. A. Este`ve, M. Djafari Rouhani et al, J. Chem. Theory Comput. 2008, 4, 1915–1927 Non-ALD reaction (MD superior to optimization) • Ligand decomposition We find that activation energies are tuned by coordination number Rate catalogue Reactions Barrier (eV) HfX4(g)HfX4(s) - HfX4(s)HfX3(s) 2.80 HfX3(s)HfX2(s) 2.96 Densification 0<0.5 HfX2(s)HfX1(s) 1.29 HfX1(s)HfX0(s) 1.64 HfX0(s)HfX1(s) 0.69 HfX1(s)HfX2(s) 2.24 HfX2(s)HfX3(s) 5.26 HfX3(s)HfX4(s) 4.92 Ligand decomposition <0.5 Ligand transfer <0.5 H2OOH-+H+ Depends on coordination number of hafnium atoms at surface H+ diffusion to amide group 0-2eV Outline • Atomic layer deposition • Findings from atomic-scale modelling • Strategy for Kinetic Monte Carlo1 • Call reaction catalogue • Stick to on-site KMC • Tie rates to coordination number of atoms at surface • Implementation of new application into the SPPARKS2 code in progress 1. 2. Arthur F. Voter, Introduction to the Kinetic Monte Carlo Method SPPARKS http://www.sandia.gov/~sjplimp/spparks.html BKL algorithm1 1. A. Bortz, M. Kalos, and J. Lebowitz, J. Comput. Phys. 17, 10 1975 Conclusions • New mechanisms of ALD reactions were found and quantified. • Ab initio MD superior to optimisation methods in identifying global basins. • Role of coordination number is important in growth of complex material. • Introduce new application for KMC. Acknowledgement We are grateful for funding by Science Foundation Ireland under the FORME project, http://www.tyndall.ie/forme/ and acknowledge a generous grant of computing time from the SFI and HEA-funded Irish Centre for High End Computing (ICHEC). We also thank A. Esteve & M. D. Rouhani in LAAS and Steve Plimpton & Corbett Battaile at Sandia National Laboratory for their collaboration. Thank you for your attention