Programmed Cell Death
advertisement

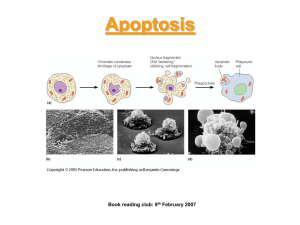
Programmed Cell Death Why do some cells need to die? • To accomplish morphogenetic ends, eg. Separation of fingers during embryogenesis of the hand • To eliminate one potential pathway of bipotential tissues and organs, eg. Elimination of Mullerian system through Mullerian Inhibiting Factor in male sexual differentiation • To accomplish metamorphosis, eg. Loss of larval structures in insect metamorphosis • To eliminate self-recognizing B-cell clones in development of vertebrate immune system • To eliminate senescent cells in organs that continuously renew themselves, eg. The intestinal epithelium and other ectodermal structures. • To eliminate cells that have begun to behave abnormally, eg. Effect of tumor necrosis factor on tumors There are at least 3 distinct forms of programmed cell death • Apoptosis or type 1 cell death – Major effect is through nuclear damage and chromosome fragmentation – but mitochondrial damage is also typical – This is the best understood form • Autophagic cell death or type 2 cell death – Major effect is through formation of autolysosomes – the cell digests itself • Type 3 cell death – Empty spaces appear in the cytoplasm How is programmed cell death initiated? • Possibility #1: cells self-initiate a suicide program unless they receive deathsuppressing growth factors. • Possibility #2: an extrinsic signal initiates the death program in cells that would otherwise survive. Nerve growth factor – an example of a system that follows possibility #1 • Viktor Hamburger and Rita Levi-Montalcini observed that growth and survival of embryonic motor neurons is dependent on the presence and size of their target tissues. Specifically, removing a limb bud from a chick embryo results in a reduction in the number of spinal motor neurons and sensory neurons in the corresponding spinal segments, whereas transplantation of an extra limb bud increased the number of neurons. • The mediator of this is secretion of trophic factors by the targets – including nerve growth factor or NGF. NGF improves neuron survival and stimulates growth of processes that will ultimately become axons. It is a member of the family of neurotropins. C. elegans – one of the simplest animals with a nervous system Nervous system development in the nematode Caenorhabditis elegans: an example of Possibility #2 • During neurogenesis, exactly 1030 neurons are born – and exactly 131 of them die. • Timing and location of neuronal apoptosis is predictable in each case. • Two genes – CED-3 and CED-4 – are required for apoptosis – site-directed mutations that inactivate either one prevent all developmentally related neuron death. • The product of CED-3 is a protease that is activated by the product of CED-4 during apoptosis. How is CED-3 turned on? • A 3rd gene CED-9 produces a product that blocks the effect of CED-4. Mutation of CED-9 results in death of cells that would otherwise survive, so the animals carrying this mutation have short lives. • CED-9 is controlled by expression of EGL-1 – the EGL-1 gene product binds to the CED-9 gene product, inactivating it. • These findings triggered a search for homologs of the CED genes in mammals, and led to the discovery of caspases. Caspases and apoptosis • Apaf-1 is the mammalian homolog of CED-4 – so this is a proapoptotic gene. • Caspases are the homologs of CED-3 • Bcl-2 proteins are the homologs of CED-9 The caspase cascade • In healthy cells, caspases are expressed as procaspases - zymogens, proteins that have to be cleaved into specific pieces to become active enzymes. • There are two classes of caspases, initiators and effectors (or executioners) – Initiators, once activated, cleave executioners – Executioners attack a wide variety of cytoplasmic and nuclear proteins How is the caspase cascade turned on? • The extrinsic pathway is turned on, for example, by ligand binding to a cell surface receptor – several death receptors have been characterized extensively. For example, one receptor for NGF kills the neurons that have it, rather than stimulating them. • The intrinsic pathway is turned on by mitochondrial release of proapoptoptic factors, such as cytochrome c. • Proteins in the Bcl-2 family (there are about 20 of them in the human genome) reside in the mitochondrial inner membrane and can prevent this release, or promote it, depending on which particular subdomains they possess. Obviously, the caspase cascade involves a number of players. To make some generalizations about it: 1. Caspases 3, 6 and 7 form a common pathway that leads to cleavage of DNA and nuclear damage 2. Several cell-surface receptors can address caspaces 3, 6 and 7, including tumor necrosis factor TNF, the Fas ligand FasL and growth factor receptor binding protein GRB. 3. A separate pathway leads from mitochondrial damage that releases Cyt C, to activation of caspases 3,6, and 7. Apoptosis and disease • Failure of apoptosis: autoimmune diseases like lupus erythematosis; some forms of cancer (so genes that regulate apoptosis are protooncogenes – mutation of them can cause cancer. • Premature or excessive apoptosis: neurodegenerative diseases such as Parkinson’s D. or amyotrophic lateral sclerosis; excitotoxicity in the nervous system. Amyotrophic lateral sclerosis • Results from apoptosis of spinal motor neurons • Associated with a mutation in superoxide dismutase 1, a cytoplasmic enzyme that scavenges oxygen free radicals, so “oxidative stress” can initiate apoptosis. If the mutant human gene is transferred to mice, they get a disease similar to the human one. • Overexpression of an antiapoptotic Bcl-2 or administration of caspase inhibitors can slow progression of the disease in the animal model. Molecular Pharmacology • A majority of cancer chemotherapy drugs act on some aspect of apoptosis. • Trials of antisense RNAs directed against antiapoptotic pathways are in progress.