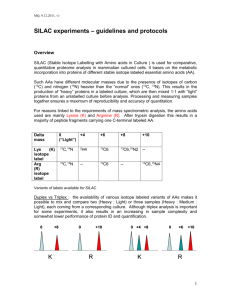
Label-free
advertisement

Quantitative Proteomics: Applications and Strategies Gustavo de Souza IMM, OUS October 2013 A little history… 1985 – First use: up to a 3 kDa peptide could be ionized 1987 – Method to ionize intact proteins (up to 34 kDa) described Instruments have no sequence capability 1989 – ESI is used for biomolecules (peptides) Sequence capability, but low sensitivity 1994 – Term «Proteome» is coined 1995 – LC-MS/MS is implemented «Gold standard» of proteomic analysis 2DE-based approach 2DE-based approach “I see 1000 spots, but identify 50 only.” LC-MS Column (75 mm)/spray tip (8 mm) Reverse-phase C18 beads, 3 mm No precolumn or split 15 cm Sample Loading:500 nl/min Gradient elution:200 nl/min ESI Platin-wire 2.0 kV Fenn et al., Science 246:64-71, 1989. MS-based quantitation Inlet LC Ion Source Mass Analyzer MALDI ES Time-of-Flight Quadrupole Ion Trap Quadrupole-TOF Peak intensities can vary up to 100x between duplicate runs. Detector Quatitative analysis MUST be carried on a single run. Ion Intensity = Ion abundance MS measure m/z Sample 2 Intensity Sample 1 m/z Isotopic Labeling Unlabeled peptide: a) b) Element 1 H Labeled peptide: a) b) Stable Isotope 2 H 12C 13C 14 15 N 16O N 18O Enzymatic Labeling Metabolic Labeling SILAC Cells in normal culture media Media with Normal AA () Media with Labelled AA (*) Start SILAC labelling by growing cells in labelling media m/z m/z (labelled AA / dialized serum) * m/z Passage cells to allow incorporation of labelled AA m/z * m/z By 5 cell doublings cells have incorporated m/z * X3 m/z Grow SILAC labelled cells to desired number of cells for experiment X3 m/z Ong SE et al., 2002 Chemical Labeling ICAT Reagents: Heavy reagent: d8-ICAT (X=deuterium) Light reagent: d0-ICAT (X=hydrogen) O N N S Biotin tag O XX N O XX O XX O XX Linker (heavy or light) O N I Thiol specific reactive group Gygi SP et al., 1999 ICAT (Isotope-Coded Affinity Tag) ICAT Cell State 1 (All cysteines labeled with light ICAT) ICAT) Combine Optional fractionation Pr ot ei n A Pr ot ei n B Pr ot ei n C Pr ot ei n D Pr ot ei n E Pr ot ei n F Relative Abundance 100 .... 0 Time Quantitate relative protein levels by measuring peak ratios Proteolyze Identify proteins by sequence information (MS/MS scan) Cell State 2 (All cysteines labeled with heavy ICAT) Analyze by LCLC-MS/MS Relative Abundance 100 Affinity separation =Protein A NH2-EACDPLREACDPLR-COOH 0 200 400 600 m/z Thiol-specific group = binds to Cysteins 800 ICAT Thiol-specific group = binds to Cysteins Intensity Quantitation at MS1 level m/z Double sample complexity, i.e. instrument have more “features” to identify, i.e. decrease in identification rate iTRAQ (isobaric Tag for Relative and Absolute Quantitation) Recognizes Arg or Lys Total mass of label = 145 Da ALWAYS Sample prep iTRAQ iTRAQ Multiplexing Metabolic VS Chemical Labeling • Metabolic labeling - 15N labeling - SILAC • Chemical methods - many… but ICAT is prototype Living cells Efficient labeling Simple! Isolated protein sample Depends on chemistry Multi-step protocols Require optimization Summary Kolkman A et al., 2005 Label-free Mobile phase C18 column, 25cm long A B 20 s Time A = 5% organic solvent in water B = 95% organic solvent in water Label-free Strassberger V et al., 2010 Summary Summary Take home message 1. Quantitation can be done gel-free 2. Labeling can be performed at protein or peptide level, during normal cell growth or in vitro 3. Quantitation can be achieved at MS1 or MS2 level 4. Method choice depends on experimental design, costs, expertise etc 5. In my PERSONAL OPINION, chemical label should be avoided at all costs unless heavy multiplexing is required Applications State A State B Upregulated protein - Peptide ratio >1 Arg13C 6 Light Isotope Heavy Isotope Mix 1:1 Arg12C 6 Optional Protein Fractionation m/z Digest with Trypsin Control vs Tumor Cell? Protein Identification and Quantitation by LC-MS Control vs drug treated cell? Control vs knock-out cell? Applications – Cell Biology Geiger T et al., 2012 Applications – Cell Biology Applications – Immunology Meissner et al, Science 2013 Clinical Proteomics A. Amyloid tissue stained in Congo Red; B. After LMD. Wisniewski JR et al., 2012 Interactomics Schulze and Mann, 2004 Schulze WX et al., 2005 Signaling Pathways Take home message 1. Anything is possible! SILAC Gustavo de Souza IMM, OUS October 2013 SILAC Cells in normal culture media Media with Normal AA () Media with Labelled AA (*) Start SILAC labelling by growing cells in labelling media m/z m/z (labelled AA / dialized serum) * m/z Passage cells to allow incorporation of labelled AA m/z * m/z By 5 cell doublings cells have incorporated m/z * X3 m/z Grow SILAC labelled cells to desired number of cells for experiment X3 m/z Ong SE et al., 2002 Importance of Dialyzed Serum • non-dialzed serum contains free (unlabeled) amino acids! No alterations to cell phenotype C2C12 myoblast cell line Labeled cells behaved as expected under differentiation protocols Why SILAC is convenient? Why SILAC is convenient? • Convenient - no extra step introduced to experiment, just special medium • Labeling is guaranteed close to 99%. All identified proteins in principle are quantifiable • Quantitation of proteins affected by different stimuli, disruption of genes, etc. • Quantitation of post-translational modifications (phosphorylation, etc.) • Identification and quantitation of interaction partners Catch 22 - SILAC custom formulation media (without Lys and/or Arg) $$$$$$ - Labeled amino acids – Lys4, Lys6, Lys8, Arg6, Arg10. Use formulation accordingly to media formula (RPMI Lys, 40mg/L) ***** When doing Arg labeling, attention to Proline conversion! (50% of tryptic peptides in a random mixture predicted to contain 1 Pro) Proline Conversion! Typical SILAC experiment workflow State A State B Upregulated protein - Peptide ratio >1 Arg13C 6 Light Isotope Heavy Isotope Mix 1:1 Arg12C 6 Optional Protein Fractionation m/z Background protein - Peptide ratio 1:1 Digest with Trypsin Arg12C 6 Arg13C 6 Protein Identification and Quantitation by LC-MS m/z Additional validation criteria * Never use labelled Arg or Lys with same mass difference (Lys6/Arg6) Triple SILAC Triple Encoding SILAC allows: 3 Intensity 2 Monitoring of three cellular states simultaneously Study of the dynamics of signal transduction cascades even in short time scales m/z Blagoev B et al., 2004 Five time-point “multiplexing” profile Blagoev B et al., 2004 Quantitative phosphoproteomics in EGFR signaling 8x 0’ EGF 8x 1’ EGF 0-5-10 min. 8x 5’ EGF Cytoplasmic ext. Nuclear extract 8x 5’ EGF 8x 10’ EGF 8x 20’ EGF SILACHeLa cells 1-5-20 min. Cytoplasmic ext. Nuclear extract Lysis and Fractionation Anf digestion SCX / TiO2 SCX / TiO2 SCX / TiO2 4x (10 SCX-fractions +FT) 44 LC-MS runs SCX / TiO2 Phosphopeptide enrichment ID and quantitation Blagoev B et al., 2004 MAP kinases activation Signal progression 40 EGFr-pY1110 ShcA-pY427 ERK1-pY204 ERK2-pY187 EMS1-pS405 10 2 1 5 10 15 EGF (minutes) 20 Spatial distribution of phosphorylation dynamics Cytosolic STAT5 translocates to the nucleus upon phosphorylation Interactomics Schulze and Mann, 2004 Schulze WX et al., 2005 Limitations - Expensive - Quantitation at MS1 level increased sample complexity - Cells has to grow in culture. Not a choice for primary cells, tissues or body fluids. - Cell lines have to be dyalized serum-friendly. SILAC-labeled organism Sury MD et al., 2010 Super-SILAC Geiger T et al., 2010 Spike-In SILAC Geiger T et al., 2013 Take home message 1. Arguably the best labeling strategies: easy to handle, no chemical steps, >98% incorporation low variability 2. Successfully used in the most diverse applications 3. Cells must be stable and growing in the media 4. There are decent alternative strategies for primary cells or organisms. Label-free Gustavo de Souza IMM, OUS October 2013 Label-free Label-free 10 s 500 fmol peptide Time 100 fmol peptide Time Strassberger V et al., 2010 Label-free Kiyonami R. et al, Thermo-Finnigan application note 500, 2010. Measurements Label-free Ideal (low std) x x x x x x Measurements Replicates Reality (late 90’s) x x x x x x Replicates Label-free Strassberger V et al., 2010 Label-free Neilson et al., Proteomics 2011 Spectral Count MS1 (or MS) 899.013 899.013 MS2 (or MS/MS) 899.013 Spectral Count 20 s Time Time Depending on how complex the sample is at a specific retention time, the machine might be busy (i.e., doing many MS2) or idle (i.e., few or none MS2) Limitation in Spectral Count MS scan MS2 scan 2 counts Time 2 counts Time Area Under Curve measurement Ion Intensity AUC Retention Time Area Under Curve measurement Ion intensity in one MS1 MS2 scan Retention Time Importance of Resolution for label-free 2+ 2+ 3+ 3+ RT RT m/z m/z Importance of Resolution for label-free -Label-free became reliable (*) Cox and Mann, Nature Biotechnol 26, 2008. Area Under Curve measurement AV: 1 NL: 3.43E6 798.32 798.83 2 1 799.33 3 799.83 82 794 x 795.17 795 802.13 797 1. Retention time 2. Peak intensity 3. Monoisotopic mass accuracy 803.40 797.73 796.31 796 800.32 802.72 798 799 m/z 800 801 802 803 805.84 804 805 Cox and Mann, Nature Biotechnol 26, 2008. Regarding Label-free… - Calculate individual peptide “Intensity”. Protein Intensity = mean of peptides intensities - LFQ normalization Data without Normalization -7422 proteins identified - 7105 proteins quantified (95.72%) How this was demonstrated? Yeast model Ghaemmagami S. et al., Nature 425, 2003 Huh WK. et al., Nature 425, 2003 How this was demonstrated? Ghaemmagami S. et al., Nature 425, 2003 MaxQuant and Yeast De Godoy LM. et al, 2008. -Label-free became reliable AND showed good correlation with a well-established model Label-free in primary cells Higher CD4+ Higher CD8+ Pattern Recognition Receptors Pathway Label-free in primary cells Infection with Sendai virus (activate RIG-I PRR) RIG-I knockout Take home message 1. “Labe-free” represents a myriad of ANY method that does not use any labeling 2. Area Under Curve calculations are the most appropriate 3. Reliability is heavily dependent in good instrumentation and good bioinformatics (MaxQuant) 4. Currently, almost as good as SILAC (yet slightly less accurate) SRM / MRM Gustavo de Souza IMM, OUS October 2013 A little history… So far, ID everything we can Mobile phase C18 column, 25cm long A 20 s Time B Targeted analysis In some cases, the researcher don’t want the MS instrument to waste time trying to sequence as much as possible, but just to “search” and sequence pre-determined peptides. -Biomarker research -Tracking specific metabolic pathways -Tracking low abundant proteins in challenging sample (f.ex., in serum) Plasma dynamic range Schiess R et al., 2009 Improving detection through tergeting Michalski A et al., 2011 Biomarker Discovery phase Screening the sample gives you the following info: -For protein X most intense peptides (not all peptides from same protein have the same intensity) - most common m/z format (+2, +3, PTM?) - their Retention times - their fragmentation profiles (does the +2 fragments well?) Biomarker Shorter gradient = More complex MS1 As you decrease separation resolution, you increase the chance that two or more peptides with different sequences BUT very close m/z elutes at the same time. SRM (Selected Reaction Monitoring) Different transitions from same peptide Performance with synthetic peptides Shorter gradient = More complex MS1 As you decrease separation resolution, you increase the chance that two or more peptides with different sequences BUT very close m/z elutes at the same time. Number of biomarkers discovered so far by MS 0 Spiking sinthetic labeled peptide for absolute quantitation Applying SRM to a proper model Bacterial genomic structure - 700-6000 genes - No alternative splicing - Limited PTM presence Discovery Phase Validation on metabolic network Validation on metabolic network - It open possibilities to study molecular function implications at metabolic level. - Generate knockout, discovery phase to visualize pahways possibly altered by the KO, targeted the candidate pathways for in-depth quantitation. Take home message - Targeted analysis: ignore whole sample and focus in few protein. - 1st step is to make the regular analysis to collect acquisition features for as many peptides as possible. - Relevant in Biomarker research - Very challenging for complex samples, very powerful for simpler organisms and for pure biology projects.