Comparison of NMR and X-ray Structures - Bio 5068
advertisement

NMR in biology: Structure, dynamics
and energetics
Gaya Amarasinghe, Ph.D.
Department of Pathology and Immunology
gamarasinghe@path.wustl.edu
CSRB 7752
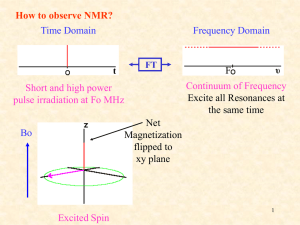
NMR?
Nuclear Magnetic Resonance
Spectroscopy
Today, we will look at how NMR can provide insight in
to biological macromolecules. This information often
compliment those obtained from other structural
methods.
NMR Spectra contains a lot of useful information:
from small molecule to macromolecule.
• Few peaks
• Sharper lines
• Overall very easy to interpret
http://www.cryst.bbk.ac.uk/PPS2/projects/schirra/html
/1dnmr.htm
• Many peaks
• Broader lines
• Overall NOT very easy to interpret
http://www.nature.com/nature/journal/v418/n6894/fig_tab/
nature00860_F1.html
• Structure determination by NMR
• NMR relaxation– how to look at
molecular motion (dynamics by
NMR)
• Ligand binding by NMR – Energetics
Outline for Bio 5068
December 11
• Why study NMR (general discussion)
1.What is the NMR signal (some theory)
2.What information can you get from NMR (structure, dynamics, and energetic
from chemical shifts, coupling (spin and dipolar), relaxation—next class)
3.What are the differences between signal from NMR vs x-ray crystallography
(we will come back to this after going through how to determine structures by
NMR)
• Practical aspects of NMR
1.instrumentation
2.Sample signal vs water signal
3.Sample preparation (very basic aspects & deal with specific labeling during
the description of experiments)
• Assignments and structure determination
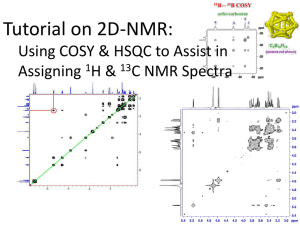
1.2-D experiments
2.3/4-D experiments
3.Restraints and structure calculations
• Assessing quality of structures
1.NMR structure quality assessment
2.Comparison with x-ray
Diffractions
For diffraction, the limit
of resolution is ½
wavelength!!
Electronic transitions
Translational transitions
Rotational transitions
Nuclear transitions
NMR works in the rf rangeafter absorption of energy by nuclei,
dissipation of energy and the time it takes
Reveals information about the conformation
and structure.
Protein Structures from an NMR Perspective
Background
–
We are using NMR Information to
“FOLD” the Protein.
–
We need to know how this NMR data
relates to a protein structure.
–
We need to know the specific details of
properly folded protein structures to
verify the accuracy of our own
structures.
–
We need to know how to determine
what NMR experiments are required.
–
We need to know how to use the NMR
data to calculate a protein structure.
–
We need to know how to use the
protein structure to understand
biological function
Protein Structures from an NMR Perspective
Analyzing NMR Data is a Non-Trivial Task!
there is an abundance of data that needs to be interpreted
X
Not A Direct Path!
Interpreting NMR Data Requires
Making Informed “Guesses” to
Move Toward the “Correct” Fold
Distance from Correct Structure
Initial rapid convergence to
approximate correct fold
Correct structure
NMR Data Analysis
Iterative “guesses” allow
“correct” fold to emerge
Current PDB statistics (as of 3/27/2012)
Exp.Met
Nucleic Protein/Nucleic
Proteins
Total
hod
Acids Acid Complexes
X-RAY
65828
1346
3260
70436
NMR
8167
975
186
9335
ratio
8.06
1.38
17.53
Nuclei are positively charged
many have a spin associated with them.
Moving charge—produces a magnetic field that has a magnetic moment
Spin angular moment
Mass
Charge
I
Even
Even
I=0
Even
Odd
I= integer
Odd
I=half integer
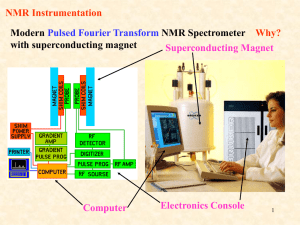
How do we detect the NMR signal?
Next time—pick up on chemical shifts
•Practical aspects of NMR
1.instrumentation
2.Sample signal vs water signal
3.Sample preparation (very basic aspects & deal with
specific labeling during the description of experiments)
http://chem4823.usask.ca/nmr/magnet.html
http://en.wikipedia.org/wiki/Nuclear_magnetic_resonance
•Practical aspects of NMR
1.instrumentation
2.Sample signal vs water signal
3.Sample preparation (very basic aspects & deal with
specific labeling during the description of experiments)
http://www.chemistry.nmsu.edu/Instrumentat
ion/NMSU_NMR300_J.html
Sample preparation using recombinant methods
Cell-free protein production and labeling protocol for NMR-based structural proteomics
Vinarov et al., Nature Methods - 1, 149 - 153 (2004)
Segment labeling can simplify NMR spectra
Native chemical ligation
Expressed protein ligation
Muir et al. Curr Opin Biotechnol. 2002 Aug;13(4):297-303.
Sample requirements and sensitivity
Methyl groups are more sensitive than isolated Ha spins
Source : www.chem.wisc.edu/~cic/nmr/Guides/Other/sensitivity-NMR.pdf
Sample requirements and sensitivity
mM not mM!!
Cryoprobes are 3-4 times better S/N than standard probes (2x in high salt)
Source : www.chem.wisc.edu/~cic/nmr/Guides/Other/sensitivity-NMR.pdf
Why use NMR ?
Some proteins do not crystallize (unstructured,
multidomain)
crystals do not diffract well
can not solve the phase problem
Functional differences in crystal vs in solution
can get information about dynamics
Protein Structures from an NMR Perspective
Overview of Some Basic Structural Principals:
a)
Primary Structure: the amino acid sequence arranged from the amino (N)
terminus to the carboxyl (C) terminus polypeptide chain
b)
Secondary Structure: regular arrangements of the backbone of the
polypeptide chain without reference to the side chain types or conformation
c)
Tertiary Structure: the three-dimensional folding of the polypeptide chain to
assemble the different secondary structure elements in a particular
arrangement in space.
d)
Quaternary Structure: Complexes of 2 or more polypeptide chains held
together by noncovalent forces but in precise ratios and with a precise
three-dimensional configuration.
Protein Structure Determination
by NMR
•Stage I—Sequence specific resonance
assignment
•State II – Conformational restraints
•Stage III – Calculate and refine structure
Resonance assignment strategies by NMR
Illustrations of the Relationship Between MW, tc and T2
NMR Assignments
3D NMR Experiments
• 2D 1H-15N HSQC experiment
• correlates backbone amide 15N through one-bond coupling to amide 1H
• in principal, each amino acid in the protein sequence will exhibit one peak in the 1H-15N
HSQC spectra
also contains side-chain NH2s (ASN,GLN) and NeH (Trp)
position in HSQC depends on local structure and sequence
no peaks for proline (no NH)
Side-chain NH2
NMR Assignments
3D NMR Experiments
• Consider a 3D experiment as a collection of 2D experiments
z-dimension is the 15N chemical shift
• 1H-15N HSQC spectra is modulated to include correlation through
coupling to a another backbone atom
C i-1
O
N i-1
C i-1
C i-1
H
H
C i
O
Ni
Ci
Ci
H
H
• All the 3D triple resonance experiments are then related by the
common 1H,15N chemical shifts of the HSQC spectra
• The backbone assignments are then obtained by piecing together all the
“jigsaw” puzzles pieces from the various NMR experiments to reassemble
the backbone
NMR Assignments
3D NMR Experiments
• Amide Strip
3D cube
2D plane
amide strip
Strips can then be arranged in backbone sequential order to visual confirm assignments
NMR Assignments
3D NMR Experiments
• 3D HNCO Experiment
common nomenclature letters indicate the coupled backbone atoms
i
i-1 (carbonyl carbon, CO or C’)
correlates NH to C
no peaks for proline (no NH)
• Like the 2D 1H-15N HSQC spectra, each amino acid should display a single peak in
the 3D HNCO experiment
1 15
identifies potential overlap in 2D H- N HSQC spectra, especially for larger
MW proteins
most sensitive 3D triple resonsnce experiment
may observe side-chain correlations
C i-1
O
C i
O
Ci
Ci
1J
NC’
N i-1
C i-1
C i-1
Ni
1J
NH
H
H
H
H
NMR Assignments
3D NMR Experiments
• 3D HN(CA)CO Experiment
correlates NHi to COi
i
relays the transfer through C without chemical shift evolution
uses stronger one-bond coupling
contains only intra correlation
provides a means to sequential connect NH and CO chemical shifts
i
i
i
i-1 (HNCO)
match NH -CO (HN(CA)CO with NH -CO
not sufficient to complete backbone assignments because of overlap and
missing information
every possible correlation is not observed
need 2-3 connecting inter and intra correlations for unambiguous
assignments
no peaks for proline (no NH) breaks assignment chain
but can identify residues i-1to prolines
C i-1
C i
O
1J
NC
N i-1
C i-1
C i-1
1J
CC’
Ni
Ci
H
H
1J
NH
H
H
O
Ci
NMR Assignments
3D NMR Experiments
• 3D HN(CA)CO Experiment
Connects HNi-COi
with HNi-COi-1
HNCO and HN(CA)CO pair
for one residues NH
Amide “Strips” from the 3D HNCO and HN(CA)CO experiments
arranged in sequential order
Journal of Biomolecular NMR, 9 (1997) 11–24
NMR Assignments
4D NMR Experiments
• Consider a 4D NMR experiment as a
collection of 3D NMR experiments
still some ambiguities present when
correlating multiple 3D triple-resonance
experiments
4D NMR experiments make definitive
sequential correlations
increase in spectral resolution
– Overlap is unlikely
loss of digital resolution
– need to collect less data points for
the 3D experiment
– If 3D experiment took 2.5 days,
then each 4D time point would be a
multiple of 2.5 days i.e. 32 complex
points in A-dimension would require
an 80 day experiment
loss of sensitivity
– an additional transfer step is
required
– relaxation takes place during each
transfer
Get less data that is less ambiguous?
NMR Assignments
NMR Assignments
Why use deuteration?
• What are the advantages?
• What are the disadvantages?
Effects of Deuterium Labeling
only 15N labeled
2D 15N-NH HSQC spectrum of the 30
kDa N-terminal domain of Enzyme I
from the E. coli
15N, 2H
labeled
Current Opinion in Structural Biology 1999, 9:594–601
Protein Structure Determination
by NMR
•Stage I—Sequence specific resonance
assignment
•State II – Conformational restraints
•Stage III – Calculate and refine structure
NMR Structure Determination
With The NMR Assignments and Molecular Modeling Tools in Hand:
• All we need are the experimental constraints
Distance constraints between atoms is the primary structure determination factor.
Dihedral angles are also an important structural constraint
What Structural Information is available
from an NMR spectra?
How is it Obtained?
How is it Interpreted?
NMR Structure Determination
NOE
NOE
- a through space correlation (<5Å)
4.1Å
- distance constraint
2.9Å
Coupling Constant (J)
- through bond correlation
J
NH
CH
- dihedral angle constraint
Chemical Shift
- very sensitive to local changes
in environment
- dihedral angle constraint
Dipolar coupling constants (D)
- bond vector orientation relative
to magnetic field
- alignment with bicelles or viruses
CH
D
NH
NMR Structure Determination
Protein Secondary Structure and Carbon Chemical Shifts
1
IV
2
III
3 I
II
4
NMR Structure Determination
Protein Secondary Structure and Carbon Chemical Shifts
• TALOS +
NMR Structure Determination
Protein Secondary Structure and Carbon Chemical Shifts
• TALOS+
Given the C, C Chemical shift assignments and primary sequence
Compares the secondary chemical shifts against database of chemical shifts and
associated high-resolution structure
comparison based on “triplet” of amino acid sequences present in database
structures with similar chemical shifts and secondary structure
Provides potential f , y backbone torsion constraints
Issues: May not provide a unique solution, two or more sets of f , y are possible. Can not
initially use TALOS results if ambiguous. Can add constraint latter if consistent with
structure.
NMR Structure Determination
Protein Secondary Structure and 3JHN
• Karplus relationship between f and 3JHN
f =180o 3JHN = ~8-10 Hz -strand
f = -60o 3JHN = ~3-4 Hz -helix
Vuister & Bax (1993) J. Am.Chem. Soc. 115:7772
NMR Structure Determination
Protein Secondary Structure and 3JHN
• Karplus relationship between f and 3JHN
Measure 3JHN for a protein using HNHA
Ratio of cross-peak to diagonal intensity
yields coupling constant
Common approach to measure coupling
constants in complex protein NMR spectra
J. Am. Chem. Soc. 1993,115, 7772-7777
Protein Structure Determination
by NMR
•Stage I—Sequence specific resonance
assignment
•State II – Conformational restraints
•Stage III – Calculate and refine structure
Protein Structures from an NMR Perspective
What Information Do We Know at the Start of Determining A
Protein Structure By NMR?
Effectively Everything We have Discussed to this Point!
The primary amino acid sequence of the protein of interest.
► All the known properties and geometry associated with each
amino acid and peptide bond within the protein.
► General NMR data and trends for the unstructured (random
coiled) amino acids in the protein.
The number and location of disulphide bonds.
► Not Necessary can be deduced from structure.
Double the nOe restraints
From above
7 restraints/residue
10 restraints/residue
13 restraints/residue
16 restraints/residue
Wüthrich et al. , J. Virol. February 15, 2009; 83:1823-1836
Analysis of the Quality of NMR Protein Structures
With A Structure Calculated From Your NMR Data, How Do You Determine the
Accuracy and Quality of the Structure?
• Consistency with Known Protein Structural Parameters
bond lengths, bond angles, dihedral angles, VDW interactions, etc
all the structural details discussed at length in the beginning
• Consistency with the Experimental DATA
distance constraints, dihedral constraints, RDCs, chemical shifts, coupling constants
all the data used to calculate the structure
• Consistency Between Multiple Structures Calculated with the Same Experimental DATA
Overlay of 30 NMR Structures
Analysis of the Quality of NMR Protein Structures
As We have seen before, the Quality of X-ray
Structures can be monitored by an R-factor
• No comparable function for NMR
• Requires a more exhaustive analysis of NMR
structures
Analysis of the Quality of NMR Protein Structures
Root-Mean Square Distance (RMSD) Analysis of Protein Structures
• A very common approach to asses the quality of NMR structures and to determine
the relative difference between structures is to calculate an rmsd
an rmsd is a measure of the distance separation between equivalent atoms
two identical structures will have an rmsd of 0Å
the larger the rmsd the more dissimilar the structures
0.43 ± 0.06 Å for the backbone atoms
0.81 ± 0.09 Å for all atoms
Analysis of the Quality of NMR Protein Structures
Root-Mean Square Distance (RMSD) Analysis of Protein Structures
• A variety of approaches can be used to measure an RMSD
only backbone atoms
exclude disordered regions
only regions with defined secondary structure
only the protein’s active-site region
on a per-atom or per-residue basis
rmsd difference between
NMR and X-ray structure
Analysis of the Quality of NMR Protein Structures
Is the “Average” NMR Structure a Real Structure?
• No-it is a distorted structure
level of distortions depends on the similarity between the structures in the
ensemble
provides a means to measure the variability in atom positions between an
ensemble of structures
Expanded View of an “Average” Structure
Some very long,
stretched bonds
Position of atoms are so
scrambled the graphics
program does not know which
atoms to draw bonds between
Some regions of the structure
can appear relatively normal
Analysis of the Quality of NMR Protein Structures
As We Discussed Before, PROCHECK is a Very
Valuable Tool For Accessing The Quality of a
Protein Structure
Correct f, y, c1, c2 distribution
► Comparison of main chain and side-chain
parameters to standard values
►
Analysis of the Quality of NMR Protein Structures
NMR R-factor
• difference between expected and observed
NOEs
expected NOEs structure
observed NOEs NMR spectra
also includes unassigned NOEs
perfect fit would yield R = 0
• R-factors have not been readily adapted in NMR
community
affected by completeness of assignments,
peak overlap, sensitivity, noise, extent of
data (RDCs, coupling constants, etc
trends with rmsd without complications
Journal of Biomolecular NMR, 17: 137–151, 2000.
Protein Structures from an NMR Perspective
Analyzing NMR Data is a Non-Trivial Task!
there is an abundance of data that needs to be interpreted
X
Not A Direct Path!
Interpreting NMR Data Requires
Making Informed “Guesses” to
Move Toward the “Correct” Fold
Distance from Correct Structure
Initial rapid convergence to
approximate correct fold
Correct structure
NMR Data Analysis
Iterative “guesses” allow
“correct” fold to emerge
Timescales of Protein Motion
Energy landscape and dynamics
high energy barriers = slow rate
low energy barriers = fast rate
H
N
Why do proteins move?
•
Broad, shallow energy potential
– Thermal energy is sufficient for the protein to sample many different conformations
•
Change in conditions
– Interaction with a small molecule or binding partner, change in temperature, ion
concentration, etc.
– Now a different conformation is lower in energy
•
Sequence encodes both protein structure and protein flexibility
– Non-bonded interactions determine the lowest energy conformation(s)
Function
Stability
Flexibility
Sequence
Function requires
•Stability: the right chemical and
spatial features in the right place
to bind ligand, catalyze a chemical
reaction, etc.
•Flexibility: the ability to move in
order to control access in and out
of the active site and to provide
energy for chemical reactions
NMR Analysis of Protein Dynamics
Hydrogen-Deuterium Exchange
• As we saw before, slow exchanging NHs
allowed us to identify NHs involved in
hydrogen-bonds.
• Similarly, slow exchanging NHs are protected
from the solvent and imply low dynamic
regions.
• Fast exchanging NHs are accesible to the
solvent and imply dynamic residues, especially
if not solvent exposed.
Protein sample is exchanged into
D2O and the disappearance of NHs
peaks in a 2D 1H-15NH spectra is
monitored.
Protein Science (1995), 4:983-993.
NMR Analysis of Protein Dynamics
Hydrogen-Deuterium Exchange
• The observed NH intensity loss can be fit to a simple exponential to measure an
exchange rate (kex)
• These exchange rates may range from minutes to months!
NHs with long exchange rates indicate stable or low mobility regions of the
protein
NHs with short exchange rates indicate regions of high mobility in the protein
I = e
k ex t
or
I = 1e
k ex 1 t
2e
k ex 2 t
NMR Analysis of Protein Dynamics
Hydrogen-Deuterium Exchange
• As expected, majority of NHs that exhibit slow exchange rates are located in secondary
structures
• fast exchanging NHs are located in loops, N- and C-terminal regions
NMR Parameters for Protein Dynamics
•
•
•
•
Number of signals per atom
Line-widths
Hydrogen Exchange (H-D)
Hetero-nuclear {15N, 13C}
Relaxation measurements
– T1 (spin-lattice relaxation time)
– T2 (spin-spin relaxation time)
– Hetero-nuclear NOE
NMR Relaxation
After an RF pulse system needs to relax back to equilibrium condition
Related to molecular dynamics of system
may take seconds to minutes to fully recovery
usually occurs exponentially:
( n n e ) t = ( n n e ) 0 exp( t / T )
–
–
(n-ne)t displacement from equilibrium value ne at time t
(n-ne)0 at time zero
Relaxation can be characterized by a time T
–
relaxation rate (R): 1/T
No spontaneous reemission of photons to relax down to ground state
Two types of NMR relaxation processes
spin-lattice or longitudinal relaxation (T1)
spin-spin or transverse relaxation (T2)
z
Mo
B1
z
B1 off…
x
(or off-resonance)
y
z
y
Mxy w
1
T1 & T2
Mo
relaxation
y
x
NMR Relaxation
Spin-lattices or longitudinal relaxation
Relaxation process occurs along z-axis
transfer of energy to the lattice or solvent material
coupling of nuclei magnetic field with magnetic fields created by the ensemble of vibrational
and rotational motion of the lattice or solvent.
results in a minimal temperature increase in sample
Relaxation time (T1) exponential decay
Mz = M0(1-exp(-t/T1))
NMR Relaxation
Spin-Spin or Transverse relaxation
Relaxation process in the x,y plane
Related to peak line-width
–
T2 may be equal to T1, or differ by orders of magnitude
–
Inhomogeneity of magnet also contributes to peak width
T2 can not be longer than T1
No energy change
T2 relaxation
(derived from Heisenberg uncertainty principal)
NMR Relaxation
Mechanism for Spin-lattices and Spin-Spin relaxation
• Illustration of the Relationship Between MW, tc and T2
Conformational Exchange Increases
the Rate of Transverse Relaxation (R2)
in NMR Spectra
R2 = R20 + Rex
Rex depends on:
Kinetics:
kex = kA + kB
Thermodynamics:
pA*pB
Structure:
Dw
NMR Analysis of Protein Dynamics
k = p Dno2 /2(he - ho)
k = p Dno / 21/2
k = p (Dno2 - Dne2)1/2/21/2
k = p (he-ho)
k – exchange rate
n – peak frequency
h – peak-width at half-height
e – with exchange
o – no exchange
In the Absence of Chemical Exchange Magnetization
Refocuses Following a 180° Pulse
PreparationPreparationRelaxation
Frequency Labeling
Acquisition
Relaxation Due to Chemical Exchange Leads to Loss of
Transverse Magnetization
Preparation
Relaxation
Frequency Labeling
No Chemical Exchange
With Chemical Exchange
Acquisition
Increasing the Number of CPMG Pulses Can Recover
Magnetization Due to Rex
Relaxation
·
·
·
Frequency Labeling
Acquisition
R 2 (1 /s )
Preparation
Rex
R20
p u ls e ra te (1 /s )
For 2-state exchange in the ms-µs regime,
quantitative analysis can in principle yield:
pA, pB, kA, kB, Dw
Summary --- NMR relaxation/dynamics
• High sensitivity and site specific information
• may need isotopic labeling
•May require assignment of resonances
• Can help narrow construct space and identify
interfaces
•regions that interact with solvent or binding
partners
NMR Analysis of Protein-Ligand Interactions
NMR Monitors the Different Physical Properties That Exist
Between a Protein and a Ligand
NMR Analysis of Protein-Ligand Interactions
Ligand Line-Width (T2) Changes Upon Protein Binding
• As we have seen before, line-width is directly related to apparent MW
a small-molecule (~100-1,000Da) is orders of magnitude lighter than a typical protein
(10s of KDa)
a small molecule has sharp NMR line-widths (few Hz at most))
protein has broad line-widths (10s of Hz)
if a small molecule binds a protein, its line-width will resemble the larger MW protein
tc MW/2400 (ns)
+
Small molecule:
Sharp NMR lines
Broad NMR lines
Chemical exchange NMR timescales
• Slow isomerization of dimethyl amino group at low temperature produces
distinct signals for each methyl
• At increasing temperatures (faster exchange rates) peaks broaden and
eventually coalesce into one average signal
• For binding reactions, slow exchange (higher affinity) produces distinct signals for
free and bound states at intermediate titration points - follow binding reaction by
watching bound/free peak intensities grow/diminish
• Fast exchange - only one set of peaks throughout titration, shifting in proportion to
changing ratio of free:bound
Summary --- NMR ligand binding
• High sensitivity and site specific information
• may need isotopic labeling
•May require assignment of resonances
• Affinity measurements are only valid for low
affinity interactions
• Complex structures can be determined for
high affinity interactions
Comparison of NMR and X-ray
Structures
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
large ribosomal subunit X-ray structure
There is no theoretical limit to the size of the structure
that can be determined by X-ray crystallography.
Requires a crystal that diffracts!
- requires highly pure samples
- requires high solubility (~mM)
- requires high stability (crystal may take
weeks to months to form)
- requires absence of aggregation/ppt
- may requires seleno-Met labeling for
phase determination
- usually need to test 100s to 1,000s of
crystal conditions
- requires a protein that will form a crystal
(may require site-directed mutant, N-,Cterminal truncation or using sequences from
different species)
Science (2000) 289, 905-920
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
Conversely, there is a molecular-weight upper limit for NMR
structures.
molecular-weight of a protein is related to its radius
which in turn is related to the protein’s rotational
correlation time (tc) :
tc =
4p r
3
3 kT
where:
r = radius
k = Boltzman constant
= viscosity coefficient
rotational correlation time (tc) is the time it takes a
molecule to rotate one radian (360o/2p).
the larger the molecule the slower it moves
tc is related to the efficiency of T2 relaxation
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
As we have seen to this point, that an NMR
structure is determined indirectly by
combining NMR experimental data as target
functions with traditional geometrical
potential energy functions.
Conversely, an X-ray structure is determined by
directly fitting the structure against the
electron density maps. This approach still uses
XPLOR to refine the structure and maintain
proper geometry (bond lengths, bond angles)
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
As a result, a single optimal structure can be
determined to represent the experimental Xray data where the r-factor indicates the
quality of the fit and the data indicates the
resolution of the structure
The EMBO Journal (2000) 19(13) 3179
Conversely, the NMR data can be equally
represented by an ensemble of structures and
there currently is no corresponding equivalent
to the r-factor or resolution
Biochemistry (2000) 39(31), 9146-9156
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
Example of Ultra-High
Resolution X-ray Diffraction
Pattern
Resolution increases (d) as you
move out concentric circles in
the X-ray diffraction pattern
Bragg equation: 2dsinf =nl
Xfd
X
Note: diffraction intensity decreases
as you move to outer circle
Acta Cryst. (2000). D56, 1015–1016
The resolution of the structure is the minimum separation of two groups in
the electron-density plot that can be distinguished from one another.
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
NMR and X-ray structures generally exhibit the same
fold
Local differences may be attributed to:
1) dynamics
2) crystal-packing interactions
3) solid vs. solution state
- solvent is present in crystals
- lowest energy conformer in crystal?
4) Resolution/experimental error
Nevertheless, there are some examples where
distinct functional differences are observed
between the NMR and X-ray structures
Protein Science (1996). 5:2391-2398.
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
Illustration of the large differences
between the NMR (blue) and X-ray
(red) structures of the Ca2+–calmodulin
complex
X-ray structure suggested a “dumb-bell”
structure with an extended -helix
NMR structure indicated the central
helix was unstructured and dynamic.
“The difference between the crystal and solution structures of Ca2+–calmodulin
indicates considerable backbone plasticity within the domains of calmodulin, which is
key to their ability to bind a wide range of targets.”
Nature Structural Biology (2001), 8(11), 990-997.
NMR and X-ray Structures
Comparison of NMR
and X-ray Structures
Protein Dynamics Is Routinely Measured From
NMR Data
Dynamic Data Is Also Implied From the X-ray BFactor (temperature factor in the PDB).
Overall Poor Correlation Between NMR Dynamic
Data and B-factors
1) dynamic regions may have low B-factors if
stabilized by an interaction not present in
solution
2) low dynamic regions may have high Bfactors due to resolution issues not related
to dynamics – various crystal contacts, lack
of uniformity in crystals, etc.
Final thoughts?
Some Other Recommended Resources
“NMR of Proteins and Nucleic Acids” Kurt Wuthrich
“Protein NMR Spectroscopy: Principals and Practice”
John Cavanagh, Arthur Palmer, Nicholas J. Skelton, Wayne Fairbrother
“Principles of Protein Structure” G. E. Schulz & R. H. Schirmer
“Introduction to Protein Structure” C. Branden & J. Tooze
“Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis”
R. Copeland
“Biophysical Chemistry” Parts I to III, C. Cantor & P. Schimmel
“Principles of Nuclei Acid Structure” W. Saenger
Some Important Web Sites:
RCSB Protein Data Bank (PDB)
http://www.rcsb.org/pdb/
Database of NMR & X-ray Structures
BMRB (BioMagResBank)
http://www.bmrb.wisc.edu/
Database of NMR resonance assignments
CATH Protein Structure Classification Classification of All Proteins in PDB
http://www.biochem.ucl.ac.uk/bsm/cath/
SCOP: Structural Classification of Proteins Classification of All Structures into
http://scop.berkeley.edu
Families, Super Families etc.
DALI
http://www.ebi.ac.uk/dali/
Compares 3D-Stuctures of Proteins to
Determine Structural Similarities of New
Structures
NMR Information Server
http://www.spincore.com/nmrinfo/
NMR Groups, News, Links, Conferences, Jobs
NMR Knowledge Base
http://www.spectroscopynow.com/
A lot of useful NMR links
Many slides have been either taken directly or adapted from the following sources:
http://www.bionmr.com/forum/educational-web-pages-16/lectures-nmr-spectroscopyprotein-structures-university-nebraska-lincoln-324/
David Cistola (Wash U)
Kevin Gardner/Carlos Amzcua (UTSW)
Or as cited