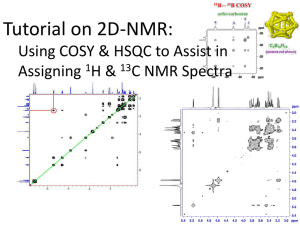
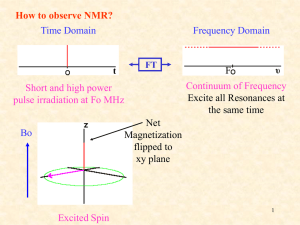
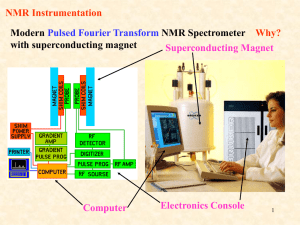
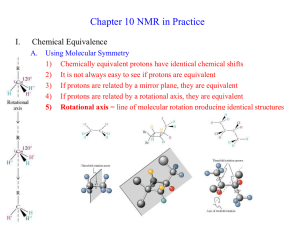
CHEM 430 * NMR Spectroscopy
advertisement

Introduction to Spectroscopy Spectroscopy is the study of the interaction of matter with the electromagnetic (EM) spectrum 1. EM radiation displays the properties of both particles and waves 2. This “packet” of wave and particle properties is called a photon The term “photon” is implied to mean a small, massless particle that contains a small wave-packet of EM radiation/light 3. The energy E component of a photon is proportional to the frequency n E = hn The constant of proportionality is Plank’s constant, h CHEM 430 – NMR Spectroscopy 2 Introduction to Spectroscopy 4. Because the speed of light (c ) is constant, the frequency (n) (number of cycles of the wave per second) can complete in the same time, must be inversely proportional to how long the oscillation is, or wavelength (l): n= c ___ l D E = hn = hc ___ l 5. Amplitude describes the wave height, or strength of the oscillation 6. Because the atomic particles in matter also exhibit wave and particle properties (though opposite in how much) EM radiation can interact with matter in two ways: • Collision – particle-to-particle – energy is lost as heat • Coupling – the wave property of the radiation matches and movement the wave property of the particle and “couple” to the next higher quantum mechanical energy level CHEM 430 – NMR Spectroscopy 3 Introduction to Spectroscopy Remember atoms and molecules are quantum mechanical particles 9. Where a photon is a wave with some particle character, matter is made of particles with some wave character – wave/particle duality 10. As a result of this, the energy of these particles can only exist at discrete energies – we say these energy levels are quantized 11. It is easy to understand if we visualize the “wave” property of matter as an oscillating string in a box—only certain “energy levels” can exist as the string is bound at both ends: CHEM 430 – NMR Spectroscopy Energy 8. 4 The Spectroscopic Process 2. Absorption: Molecule takes on the quantum energy of a photon that matches the energy of a transition and becomes excited excited state Energy hn 5. Detection: Relaxation as photons are reemitted. Spectrometers differ whether they measure actual emission or absorbance rest state hn hn hn 1. Irradiation: Molecule is bombarded with photons of various frequencies over CHEMdesired 430 – NMR Spectroscopy the range rest state 5 5 Types of Spectroscopy g-rays (X-ray cryst.) X-rays Frequency, n (Hz) Wavelength, l Energy (kcal/mol) ~1017 ~0.01 nm > 300 electronic excitation (p to p*) molecular molecular vibration rotation Visible nuclear excitation (PET) core electron excitation UV ~1015 10 nm 300-30 IR Microwave Nuclear Magnetic Resonance NMR & MRI Radio ~1013 ~1010 ~105 1000 nm 0.01 cm 100 m 300-30 ~10-4 ~10-6 CHEM 430 – NMR Spectroscopy 6 6 NMR Spectroscopy • NMR spectroscopy has emerged as the ultimate spectroscopic method for organic structural analysis • Currently, the development of novel NMR methods is in its “golden age” with some of the 2-D methods entering their maturation period as routine spectroscopic methods • A typical NMR sample consists of 1-10 mg of sample, with which a full analysis of 1H, 13C, DEPT, COSY, HMBC, HSQC and NOESY could be done in a few hours on a high-field instrument • Important spin-offs of NMR spectroscopy include a host of medical and security imaging equipment CHEM 430 – NMR Spectroscopy 7 Brief History of NMR • First NMR spectrum of H2O, 1946: Bloch, F.; Hansen, W. W.; Packard, M. Phys. Rev. 1946, 70 474-85. CHEM 430 – NMR Spectroscopy 8 Brief History of NMR • First observation of chemical shift 1H spectrum of ethanol – 1951 vs. 2011 Arnold, J.T., S.S. Dharmatti, and M.E. Packard, J. Chem. Phys., 1951, 19, 507. CHEM 430 – NMR Spectroscopy 9 Brief History of NMR • Fourier transform NMR by Ernst - 1966 CHEM 430 – NMR Spectroscopy 10 Brief History of NMR • 2D NMR – 1975 Jeener and Ernst CHEM 430 – NMR Spectroscopy 11 Brief History of NMR (MRI) • First magnetic resonance image – 1973 Lauterbur and Mansfield 2011 CHEM 430 – NMR Spectroscopy 12 Brief History of NMR • First 3-D spectrum of small protein - 1985 Wüthrich CHEM 430 – NMR Spectroscopy 13 Brief History of NMR Nobel Prizes for NMR •1944 Physics Rabi (Columbia) •1952 Physics Bloch (Stanford), Purcell (Harvard) •1991 Chemistry Ernst (ETH) •2002 Chemistry Wüthrich (ETH) •2003 Medicine Lauterbur (University of Illinois in Urbana ) and Mansfield (University of Nottingham) CHEM 430 – NMR Spectroscopy 14 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • The sub-atomic particles within atomic nuclei possess a spin quantum number just like electrons • Just as when using Hund’s rules to fill atomic orbitals with electrons, nucleons must each have a unique set of quantum numbers • The spin quantum number of a nucleus is a physical constant, I • For each nucleus, the total number of spin states allowed is given by the equation: 2I + 1 CHEM 430 – NMR Spectroscopy 15 Basis of NMR Spectroscopy Spin Quantum Numbers of Common Nuclei Element 1H 2H 12C 13C 14N 16O 17O 19F 31P 35Cl Nuclear Spin Quantum Number ½ 1 0 ½ 1 0 5/2 ½ ½ 3/2 # of spin states 2 3 0 2 3 0 6 2 2 4 • Observe that for atoms with no net nuclear spin, there are zero allowed spin states • Nuclear Magnetic Resonance can only occur where there are allowed spin states • Note that two nuclei, prevalent in organic compounds have allowed nuclear spin states – 1H and 13C, while two others do not 12C and 16O CHEM 430 – NMR Spectroscopy 16 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • There are three types of nuclei: • No spin: I = O • 12C, 16O • Cannot be observed by NMR • Spinning sphere: I = ½ • 1H, 13C, 15N, 19F, 29Si, 31P) • Easiest to observe by NMR • Spinning ellipsoid I = 1, 3/2, 2… • 2H, 11B, 14N, 17O, 33S, 35Cl • Difficult to observe by NMR CHEM 430 – NMR Spectroscopy 17 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • A nucleus contains protons, which each bear a +1 charge • If the nucleus has a net nuclear spin, and an odd number of protons, the rotation of the nucleus will generate a magnetic field along the axis of rotation m I = +½ H H I = -½ m • A hydrogen atom with its lone proton making up the nucleus, can have two possible spin states—degenerate in energy CHEM 430 – NMR Spectroscopy 18 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • The magnitude of m varies from atom to atom : m = għI • ħ is Planck’s constant divided by 2p • g is the characteristic gyromagnetic ratio of the nucleus • The larger g is, the greater the magnetic moment CHEM 430 – NMR Spectroscopy 19 Nuclear Magnetic Resonance In the absence of stimulus all nuclear spin sates are degenerate When a large magnetic field B0 is applied the two spin states become nondegenerate CHEM 430 – NMR Spectroscopy As B0 increases, the larger DE becomes 20 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • The large external magnetic field is defined as B0 in units of Tesla, T • The axis of B0 is defined as the z-direction • Splitting of spins into quantized groups is called the Zeeman effect DE direction CHEM 430 –BNMR 0 +z Spectroscopy 21 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • The force of B0 causes m to move in a circular motion about the zdirection – precession • B0 field in z-direction operates on the x component of m to create a force in the y-direction (F = m X B0) • This occurs with an angular frequency w0 known as the Larmor frequency (rad s-1) CHEM 430 – NMR Spectroscopy 22 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • A quantum mechanical particle can absorb a photon of energy equal to DE and become promoted to a higher state – spectroscopic process • As B0 increases so does w0 (B0 w); the constant of proportionality is g: w0 = g B0 • By equating w0 with Planck’s relationship: DE = hn0 = ħw0 = g B0 CHEM 430 – NMR Spectroscopy 23 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • What does this mean for the NMR experiment (observing DE)? • Solving for the frequency of EM radiation we are observing: • For a bare hydrogen nucleus (H+), g = 267.53 (106 rad/T·s) • In a B0 of 1.41 Tesla: DE = 60 MHz DE corresponds to the highly weak radio region of the EM spectrum: l > 5 meters and energies of < 0.02 cal·mol-1 • This causes technical challenges to observing NMR CHEM 430 – NMR Spectroscopy 24 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei Boltzmann distribution – more problems with NMR observation •DE at 60 MHz (DE = hn) is 2.39 x 10-5 kJ mol-1 (tiny) – thermal energy at room temperature (298 oK) is sufficient to populate both energy levels •DE is small, so rapid exchange is occurring between the two populations, but there is always a net excess of protons in the lower energy state •From the Boltzman’s Law we can calculate the population of each energy state: Nupper/Nlower = e-DE/kT = e-hn/kT @ 298 oK the ratio is 1,000,000 / 1,000,009 ! There is an excess population of 9 nuclei in the lower energy state! CHEM 430 – NMR Spectroscopy 25 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • As the applied B0 increases, exchange becomes more difficult and the excess increases: Field, B0 T Frequency MHz Excess nuclei 1.41 60 9 1.88 80 12 7.05 300 48 9.40 400 64 14.1 600 96 6. In each case, it is these few nuclei that allow us to observe NMR 7. When radio radiation is applied to a sample both transitions upward and downward are stimulated – if too much radiation is applied both states completely equilibrate – a state called saturation – no observed NMR signal CHEM 430 – NMR Spectroscopy 26 Basis of NMR Spectroscopy 2.2 Commonly Studied Nuclides CHEM 430 – NMR Spectroscopy 27 Basis of NMR Spectroscopy 2.2 Commonly Studied Nuclides • Remember that the greater DE the easier it is to detect NMR active nuclei and have greater S/N ratios: CHEM 430 – NMR Spectroscopy 28 Basis of NMR Spectroscopy 2.2 Commonly Studied Nuclides 1. Spin • • In general spin ½ nuclei are the easiest to observe Quadrapolar (I > ½) nuclei are more difficult to observe • Unique mechanism for relaxation gives very short relaxation time • Heisenberg uncertainty principle dictates: DE Dt ~ ħ • As relaxation times become very short, the uncertainty in energies becomes large and peaks broaden greatly CHEM 430 – NMR Spectroscopy 29 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei 2. Natural Abundance • Remember from Boltzman’s Law we have only a small excess of nuclei we can observe: 60 MHz, 1.41 T: 1,000,000 / 1,000,009 for 1H •1H is 99.985% of natural hydrogen - only 9 excess nuclei •Consider the excess population when the nuclei we are observing is 13C – 1.11% of natural carbon •Spin couplings between low-abundant nuclei are also hampered: • • Chance of two 1H-1H on adjacent Cs: 99.985 x 99.985 = 99.97% Chance of two 13C-13C adjacent to one another: 1.11 x 1.11 = 0.1% CHEM 430 – NMR Spectroscopy 30 Basis of NMR Spectroscopy 2.2 Commonly Studied Nuclides 3. Natural Sensitivity - g • Remember - DE = hn0 = ħw0 = g B0 NMR signal a function of only B0 and g • The larger DE the greater the excess population for observation (Boltzmann distribution) • 1H is best followed by 19F in routine observations CHEM 430 – NMR Spectroscopy 31 Basis of NMR Spectroscopy 2.2 Commonly Studied Nuclides 4. Receptivity • Mathematical product of abundance and g • Good quick measure of amenable a nuclei is for observation • Your text uses 13C as a guidepost rather than 1H • Quick survey: • • • 1H is 5680 times easier to observe than 13C 15N is 2.2% as easy to observe as 13C 19F is 4730 times easier to obersve than 13C; 31P is 2 times easier CHEM 430 – NMR Spectroscopy 32 Basis of NMR Spectroscopy 2.3 The Chemical Shift • Observation of the NMR phenomenon would be of little use if all protons resonated at the same frequency • In organic compounds protons are not bare nuclei, they are surrounded by an s-orbital of containing an e- shared with an e- in a hybridized orbital of another atom to form a covalent bond • In the presence of an external magnetic field, an induced circulation of electrons opposite to that of a proton is observed since the two are of opposite charges • This induced circulation generates a magnetic field in opposition to the applied magnetic field – a local diamagnetic current CHEM 430 – NMR Spectroscopy 33 Basis of NMR Spectroscopy 2.3 The Chemical Shift • Since the magnetic field “felt” by the proton within this electron cloud is lowered, the resonance condition frequency is also lowered • This effect of lowering the energy of transition by a cloud of electrons is called diamagnetic shielding or shielding - represented as s • The opposite effect – if electron density is removed from the vicinity of the proton is called deshielding • The actual field around the nucleus becomes B0(1 – s); substitution in the energy equation gives: CHEM 430 – NMR Spectroscopy 34 Basis of NMR Spectroscopy 2.3 The Chemical Shift • Since the magnetic field “felt” by the proton within this electron cloud is lowered, the resonance condition frequency is also lowered • This effect of lowering the energy of transition by a cloud of electrons is called diamagnetic shielding or shielding • The opposite effect – if electron density is removed from the vicinity of the proton is called deshielding DE direction CHEM 430 –BNMR 0 +z Spectroscopy 35 NMR Spectroscopy Introduction 2-6 Basis of NMR Spectroscopy • In acetic acid, the –CH3 protons are in an e- rich environment relative to the –OH proton. • Shielding of the electrons opposes B0 and therefore DE is lower than that observed for the –OH proton. Here DE is large as the full effect of B0 is felt DE –CH3 DE –OH CHEM 430 –BNMR direction 0 +z Spectroscopy 36 DE CHEM 430 – NMR Spectroscopy B0 37 Basis of NMR Spectroscopy • The effect of electrons on a 1.41 T magnetic field is negligible, but measurable • Compare the resonance frequencies for the protons in fluoromethane vs. chloromethane CH3F CH3Cl • The stronger inductive w/d of electrons by fluorine reduces the resonance frequency by 72 Hz (not MHz) compared to an operating frequency of the instrument at 60 MHz @ 1.41 T – barely 1 part per million (ppm) • Using units of 60000072 vs. 60000000 is clunky at best • There needs to be a reference “proton” by which these “chemical shifts” can be related - the best candidate would be a completely deshielded proton (H+) which does not exist in the solution phase CHEM 430 – NMR Spectroscopy 38 Basis of NMR Spectroscopy • NMR spectroscopists chose the other end of the spectrum- a proton that was more shielded than any other known proton (at the time) – those in tetramethylsilane (TMS) • The 12 chemically identical protons in TMS were used as the standard zero for an NMR spectrum • The resonance frequency of any proton to be studied (since all were less shielded) would be at parts per million of the operating frequency of the instrument greater than this zero • This allowed NMR instruments of varying field (and thus operating frequency) strengths to use the same scale • Here’s how: CHEM 430 – NMR Spectroscopy 39 Basis of NMR Spectroscopy • In an applied field of 1.41 T, the resonance frequency for a typical proton is 60 MHz, at 2.35 T it is at 100 MHz – a ratio of 5/3 • Thus, for a given proton, the shift in Hz from the TMS standard should be 5/3 greater in the 100 MHz instrument compared to the 60 MHz • Since these are simple ratios, we can simply factor out the effect of field strength by defining d, or chemical shift to be d = (shift from TMS in Hz) (spectrometer frequency in MHz) …or ppm of the instruments operating frequency CHEM 430 – NMR Spectroscopy 40 Basis of NMR Spectroscopy 90 Mhz spectrum 90 Hz 300 MHz spectrum CHEM 430 – NMR Spectroscopy 300 Hz 41 Basis of NMR Spectroscopy • In an applied field of 1.41 T, the resonance frequency for a typical proton is 60 MHz, at 2.35 T it is at 100 MHz – a ratio of 5/3 • Thus, for a given proton, the shift in Hz from the TMS standard should be 5/3 greater in the 100 MHz instrument compared to the 60 MHz • Since these are simple ratios, we can simply factor out the effect of field strength by defining d, or chemical shift to be d = (shift from TMS in Hz) (spectrometer frequency in MHz) …or ppm of the instruments operating frequency CHEM 430 – NMR Spectroscopy 42 Basis of NMR Spectroscopy • A detailed study of chemical shifts is the basis of Chapter 3 CH3 H Si O H3C C H C CH CH3 3 O C H O H S H O R H O O H O C C H 10 R R H C C Ph 15 C 9 8 7 downfield deshielded higher DE 6 5 d (ppm) CHEM 430 – NMR Spectroscopy 4 R = H or alkyl EWG 3 2 1 0.0 upfield shielded lower DE 43 Spectrometer Design Basis of NMR Spectroscopy Continuous-Wave (CW) Instrument An NMR spectrometer needs to perform several functions: • Generate a high (>1 Tesla) magnetic field to split the energy levels of the spin states enough to: – Create an excess nuclei population large enough to observe – Make the radio n that correspond to the transition be observable • Ensure that the field is homogeneous (shimming) • Vary either the applied field or the radiofrequency (RF) to observe different nuclei at their various energies of transition • Receive the faint signal of the relaxation of the excited nuclei to their ground state • Process the signal into a usable spectrum vs. a reference CHEM 430 – NMR Spectroscopy 44 Basis of NMR Spectroscopy Continuous-Wave (CW) Instrument: RF Detector RF (60 MHz) oscillator Permanent Magnet Variable magnetic field – 1.41 T ± few millionths of T CHEM 430 – NMR Spectroscopy 45 Basis of NMR Spectroscopy How it works (CW NMR): • The sample is placed in a 5 mm solution cell or tube (experimental aspects we will cover shortly) in the center of a large permanent or electromagnet • A RF oscillator coil at 90° to the sample generates a radio signal at the operating frequency of the instrument (60 MHz for a 1.41 T field) • The overall magnetic field is varied by a small electromagnet capping the poles of the larger field magnet • Remember: DE = n = (g/2p) B0, so variations of either magnetic field or frequency will cover the observed spectral width if the other is held constant • As with older dispersive IR instruments, the sweep of magnetic fields is simultaneous with the movement of the chart paper CHEM 430 – NMR Spectroscopy 46 Basis of NMR Spectroscopy How it works (CW NMR): • As a particular proton population comes into resonance, a second receiver coil at 90° to the transmitter coil will pick up the change in orientation of nuclear spin • This is recorded by the chart as a voltage response, proportional to the size of the proton population that generated the resonance • One artifact of CW instruments is that the relaxation of the protons is slower than the movement (sweep) of the chart paper • This causes the ringing effect – a decreasing oscillation of the signal after the spectrometer has moved past a given resonance • CW instruments operate by bringing each individual population of protons into resonance individually. CHEM 430 – NMR Spectroscopy 47 Basis of NMR Spectroscopy Limitations- CW NMR: • Since the spectrum is collected once, the sample must possess enough protons to give a suitable excess population that can be observed – need a concentrated sample • Due to the limitations of the relatively low magnetic field (CW instruments top out at 60-90 MHz) the coupling constants for JHH are relatively large compared to the spectral width – so only simple molecules can be observed and their structures elucidated • For nuclei of lower magnetogyric ratios, g, or natural abundance (13C most specifically) the ratio of radio noise to signal is high CHEM 430 – NMR Spectroscopy 48 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • Let’s revisit the NMR experiment we considered earlier • Recall that at even a high applied B0 only a small excess of nuclei are in the lower spin state (+ ½ ) as per the Boltzman distribution Ex: @ 7.04 T an excess of 50 spins per million • It is important to also note that 298 K imparts enough energy to the system such that all spins are interchanging rapidly: CHEM 430 – NMR Spectroscopy 49 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • So when we are discussing an NMR sample we have quadrillions of protons creating a net magnetization M in the z direction (B0) • The xy components are distributed randomly, and we can think of the rest state as M = Mz z z M y x y Bo x • M precesses at the Larmor frequency. We are using the rotating frame of reference to make the visualization easier. CHEM 430 – NMR Spectroscopy 50 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • Important note: • Each individual spin can only exist in either quantized state (± ½) separated by DE • The bulk magnetization however can exist at a continuum of states z M y Bo x CHEM 430 – NMR Spectroscopy 51 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • If a RF (w0 = w) is applied that creates a magnetic oscillation along the x-axis a torque is applied to M rotating it towards y z z Mo y B1 B1 y (or off-resonance) x Mxy x wo wo • With the application of energy, a small amount of the excess population has flipped spins. Mxy < M0 CHEM 430 – NMR Spectroscopy 52 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • If RF energy continues to be applied the excess of + ½ nuclei disappears – sample at saturation – no NMR signal can be detected • Natural mechanisms return an excess population to equilibrium • Any process that returns the z-magnetization to equilibrium with an excess of + ½ spins is called spin-lattice or longitudinal relaxation • It is usually a first-order process and with a time constant of T1 CHEM 430 – NMR Spectroscopy 53 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • Other magnetic nuclei in the NMR sample tumble, generating a field from its motion • If the field is at w, excess spin energy can pass to this motional energy as – ½ nuclei become + ½ nuclei • For this process to be effective the nuclei need to be spatially proximate to the tumbling molecule. • Most H-atoms are on the outside of molecules and have roughly equal probability to relax by this process - #1Hs ~ signal CHEM 430 – NMR Spectroscopy 54 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • C-atoms differ substantially in their contact with external molecules – T1 relaxation usually by attached protons • Therefore T1 relaxation is more efficient for a –13CH3 rather than a quaternary carbon or 13C=O. CHEM 430 – NMR Spectroscopy 55 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • T1 relaxation is responsible for generating the excess of + ½ nuclei in the first place! • When the sample is first placed in the B0 field, all spins are degenerate • Magnetization builds up as spins flip from the effect of interactions with surrounding magnetized nuclei CHEM 430 – NMR Spectroscopy 56 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR •Any process that returns xy magnetization to its equilibrium position of zero is called spin-spin or transverse relaxation •It is a first order process with time constant T2 •By definition T2 < T1 •For T2 relaxation the phases of nuclear spins must become randomized •The mechanism for this is when spin is transferred between nuclei of opposite spin CHEM 430 – NMR Spectroscopy 57 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR •When one spin goes from +1/2 to -1/2 while the other goes from -1/2 to +1/2 there is no net change in z magnetization •However switches in spin cause dephasing; as the process continues xy magnetization disappears CHEM 430 – NMR Spectroscopy 58 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR •Detection – as xy magnetization dephases the vectors begin to rotate about z in the rotating frame of reference •A second coil 90o to the transmitter coil detects the decay in the xy z y Mxy wo x Receiver coil (x) NMR signal CHEM 430 – NMR Spectroscopy 59 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR •The NMR process: CHEM 430 – NMR Spectroscopy 60 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR •Introduction to Pulse Sequences •Shorthand: 90x Mixing time Detection Pulse CHEM 430 – NMR Spectroscopy 61 Pulsed Fourier Transform (FT) Instrument First, what is a Fourier Transform? Fourier transforms interconvert mathematical functions in the frequency domain to the time domain: f(n) = ∫ f(t) e-int dt - f(t) = ½ p ∫ f(n) eint dt - For purposes of this discussion, we will black box the actual calculations and derivations of these functions, but we need to understand what they do CHEM 430 – NMR Spectroscopy 62 Spectrometer Design Pulsed Fourier Transform (FT) Instrument If we feed two simple oscillating equations into a FT, here are the results: f (t) = cos (nt) FT -n +n f (t) = sin (nt) FT +n CHEM 430 – NMR Spectroscopy -n 63 Spectrometer Design NMR Spectroscopy Pulsed Fourier Transform (FT) Instrument • In the FT instrument, all proton populations are excited simultaneously by a short, intense burst of RF energy • Due to a variation of the Heisenberg Uncertainty Principle, even if the RF generator is set at 90 MHz, if the duration of the pulse is short, the radio waves do not have time to establish a solid fundamental frequency • This can be illustrated by the following cartoon, showing the combination of a short pulse being added to a step function: on + off on off = off off tp = pulse duration CHEM 430 – NMR Spectroscopy 64 Spectrometer Design Pulsed Fourier Transform (FT) Instrument 4. If this short pulse is converted into the frequency domain by a FT: on FT off off tp n Observe that we have a continuum of frequency content centered at the operating frequency of the instrument We will talk more about the effects of pulse time and width when we discuss advanced 1-D and 2-D NMR CHEM 430 – NMR Spectroscopy 65 Spectrometer Design Pulsed Fourier Transform (FT) Instrument 5. For now, if a sample containing one unique population of hydrogens was excited over tp by a pulse, it would then “relax” back to its original spin state 6. As each nuclei relaxes it will emit RF radiation of a given frequency; since different nuclei will relax at different rates, the signal decays over time 7. This emission is recorded by the spectrometer as a free-induction decay or FID off off CHEM 430 – NMR Spectroscopy 66 Spectrometer Design Pulsed Fourier Transform (FT) Instrument 8. The actual frequency of the FID is the interference signal of the relaxing protons superimposed with the frequency of the RF source 9. Conversion of this decay signal by FT back into the frequency domain gives us the actual n of resonance for the proton being observed Pulse n Proton signal FT n time 10.Again to due Heisenburg and other factors, NMR signals are not single lines, but a Lorentzian shaped continuum of lines centered at the n of the signal CHEM 430 – NMR Spectroscopy 67 Spectrometer Design Pulsed Fourier Transform (FT) Instrument Advantages: Since all nuclei are excited and observed simultaneously, the pulse can be repeated after each relaxation period (for 1H, about 10 seconds) and the resulting signals added together Because we are observing weak radiofrequency signals in a sea of RF noise for dilute samples (or those observed once as in CW NMR) noise becomes an issue If several to hundreds of FIDs are added together, signals will tend to constructively add together and become more pronounced; since noise is random, it will tend to destructively add and become less pronounced Signal to noise ratio improves as a function of the square root of the scans (FIDs) performed: S/N = f (n) CHEM 430 – NMR Spectroscopy 68 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR •Back to Pulse Sequences •Standard 1-D observation of 1H or other nuclei 90x Detection Delay Pulse n = scans •Delay must be longer than T1 (and T2 < T1) •Scans are repeated until S/N ratio is high enough CHEM 430 – NMR Spectroscopy 69 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR •How can you know what T1 is? •We cannot measure it directly – in the z-axis •Pulse sequence is used called inversion recovery: 180y (or x) 90y tD CHEM 430 – NMR Spectroscopy 70 NMR Spectroscopy Basis of NMR Spectroscopy 2-6 2.4 Excitation and Relaxation – FT-NMR tD = 0 z z x y tD > 0 x FT y z z x y tD >> 0 x FT y z z x y x FT y CHEM 430 – NMR Spectroscopy 71 NMR Spectroscopy 2-6 Basis of NMR Spectroscopy Intensity, (I) • If we plot the intensity versus time we get the following curve: time I(t) = I * ( 1 - 2 * e - t / T1 ) • It is an exponential with a time constant equal to the T1 relaxation time. • Most 1Hs decay in less than 10 seconds – no need to run routinely CHEM 430 – NMR Spectroscopy 72 NMR Spectroscopy Basis of NMR Spectroscopy 2-6 2.4 Excitation and Relaxation – FT-NMR •How can you know what T2 is? •We cannot measure it directly like T1 •Pulse sequence is used called spin-echo sequence 90y 180y (or x) tD CHEM 430 – NMR Spectroscopy tD 73 NMR Spectroscopy Basis of NMR Spectroscopy 2-6 2.4 Excitation and Relaxation – FT-NMR •What it does: z y y tD x x x y y dephasing y tD x x 180y (or x) refocusing CHEM 430 – NMR Spectroscopy 74 NMR Spectroscopy 2-6 Basis of NMR Spectroscopy 2.4 Excitation and Relaxation – FT-NMR • If we acquire the FID right after the spin-echo sequence, the intensity of the signal after FT will only be affected by T2 relaxation and not by dephasing due to B0 imperfections. • Upon repetition for different tD values, we plot the intensity versus 2 * tD and get a graph similar to the one we got for inversion recovery, but in this case the decay rate will be equal to T2. CHEM 430 – NMR Spectroscopy 75 The Coupling Constant • Consider the spectrum of ethyl alcohol: • Why does each resonance “split” into smaller peaks? HO CH3 C H2 CHEM 430 – NMR Spectroscopy 76 The Coupling Constant • The magnetic effects of nuclei in close proximity to those being observed have an effect on the local magnetic field, and therefore DE • Two examples of close proximity are geminal and vicinal protons (homonuclear) and protons attached to 13C (heteronuclear): • On any one of the 108 of these molecules in a typical NMR sample, there is an equal statistical probability each proton is either spin + ½ or – ½ CHEM 430 – NMR Spectroscopy 77 NMR Spectroscopy 2-6 The Coupling Constant • This creates two different magnetic environments for a proton being observed – one where its neighbor is +½ the other where its neighbor is – ½ • If there is more than one proton on an adjacent carbon – all the statistical probabilities exist that each one is either + ½ or – ½ in spin • The summation of these effects over all of the observed nuclei in the sample is observed as the spin-spin splitting of resonances CHEM 430 – NMR Spectroscopy 78 The Coupling Constant • Consider 1-chloro-4-nitrobenzene If we are observing the resonance for the HA proton, in half the molecules in the sample HX would be –½ in the other half the spin would be +½ We would see the resonance of HA “split” into two different resonances (energy states): CHEM 430 – NMR Spectroscopy 79 The Coupling Constant • Consider 1-chloro-4-nitrobenzene Likewise, if we are observing the resonance for the HX proton, in half the molecules in the sample HA would be –½ in the other half the spin would be +½ We would see the resonance of HX “split” into two different resonances (energy states): CHEM 430 – NMR Spectroscopy 80 The Coupling Constant • Consider 1-chloro-4-nitrobenzene The observed 1H NMR spectrum shows two doublets, one for HA the other for HX HA and HX are described as a spin-system CHEM 430 – NMR Spectroscopy 81 NMR Spectroscopy The Coupling Constant 2-6 • The influence of neighboring spins on peak multiplicity is called spin-spin coupling, indirect coupling or J-coupling • The difference between the component peaks of a resonance is a measure of how strong the interaction is between adjacent nuclei • This difference is called the coupling constant, J, measured in Hz # of bonds separating nuclei 3J H-cis Nuclei that are coupled and stereochemistry where appropriate CHEM 430 – NMR Spectroscopy 82 NMR Spectroscopy Introduction 2-6 The Coupling Constant 60 MHz – propyl bromide • Since J is a measure of J-values are = interaction between two nuclei, it must be the same for both nuclei J-values are = • Similarly, J is independent of the applied B0 and will have the same value regardless of the field strength of the NMR 300 MHz – propyl bromide CHEM 430 – NMR Spectroscopy 83 NMR Spectroscopy Introduction 2-6 The Coupling Constant 60 MHz – propyl bromide • Since J is a measure of J-values are = interaction between two nuclei, it must be the same for both nuclei J-values are = • Similarly, J is independent of the applied B0 and will have the same value regardless of the field strength of the NMR 300 MHz – propyl bromide CHEM 430 – NMR Spectroscopy 84 NMR Spectroscopy 2-6 The Coupling Constant • Recall, we are observing the frequency (E = hn) where a proton goes into resonance Any change in B0 will cause a change in energy at which the resonance condition will occur for a proton of a given chemical shift CHEM 430 – NMR Spectroscopy 85 NMR Spectroscopy 2-6 The Coupling Constant • In solution we are not looking at a single molecule but about 108 • On some molecules the proton being observed may be next to another proton of spin + 1/2 : CHEM 430 – NMR Spectroscopy 86 NMR Spectroscopy 2-6 The Coupling Constant • On some molecules the proton being observed may be next to another proton of spin – 1/2 : CHEM 430 – NMR Spectroscopy 87 NMR Spectroscopy Introduction 2-6 The Coupling Constant • Mechanism for coupling: usually oversimplified in early studies of NMR - If molecules can rotate in solution any spatial effect of one protons magnetic field on another is averaged to zero • The most common mechanism involves the interaction of electrons along the bonding path between the nuclei. • Electrons, like protons, act like spinning particles and have a magnetic moment. The X proton ( HX) influences, or polarizes, the spins of its surrounding electrons, making the electron spins favor one Iz state very slightly. CHEM 430 88 NMR Spectroscopy Introduction 2-6 The Coupling Constant • Thus, a proton of spin +½ polarizes the electron to –½. The electron in turn polarizes the other electron of the bond, and so on, finally reaching the resonating A proton (HA). • Because J normally represents an interaction through bonds, it is a useful parameter for drawing conclusions about molecular bonding, such as bond order and stereochemistry. CHEM 430 89 The Coupling Constant • Observe what effect this has on an isolated ethyl group: • The two methylene Ha protons have three neighbors, Hb, on the adjacent methyl carbon • Each one of these hydrogens can be + ½ or – ½ , and since we are not looking at one molecule, but billions, we will observe all combinations CHEM 430 – NMR Spectroscopy 90 The Coupling Constant • The first possibility is that all three Hb protons have a + ½ spin; in this case the three protons combine to generate three small magnetic fields that aid B0 and deshield the protons – pushing part of the resonance for Ha slightly downfield (the magnetic field of a proton is tiny compared to B0) All 3 Hb protons + ½ +++ Resonance, n, in absence of coupling CHEM 430 – NMR Spectroscopy 91 The Coupling Constant • The second possibility is that two Hb protons have a + ½ spin and the third a - ½ ; in this case the two protons combine to enhance B0 and the other against it, a net deshielding; there are 3 different combinations that generate this state or 3 combinations of +½, +½ , -½ +++ or -++ +-+ ++- Resonance, n, in absence of coupling CHEM 430 – NMR Spectroscopy 92 The Coupling Constant • The third possibility is that two Hb protons have a –½ spin and the third +½; here, the two protons combine to reduce B0 and the other enforce it, a net shielding effect; there are 3 different combinations that generate this state or 3 combinations of +½, -½ , -½ +++ -++ +-+ ++- or --+ -++-- Resonance, n, in absence of coupling CHEM 430 – NMR Spectroscopy 93 The Coupling Constant • The last possibility is that all three Hb protons have a – ½ spin; in this case the three protons combine to oppose B0, a net shielding effect; there is one combination that generates this state: All 3 Hb protons -½ +++ -++ +-+ ++- --+ -++-- --- Resonance, n, in absence of coupling CHEM 430 – NMR Spectroscopy 94 The Coupling Constant • The result is instead of one resonance (peak) for Ha, the peak is “split” into four, a quartet, with the constituent peaks having a ratio of 1:3:3:1 centered at the d (n) for the resonance : -CHa2- is ‘split’ into a quartet by three adj. Hbs +++ -++ +-+ ++- --+ -++-- --- Resonance, n, in absence of coupling CHEM 430 – NMR Spectroscopy 95 NMR Spectroscopy • 2-6 The Coupling Constant Similarly, the Hb protons having two protons, on the adjacent carbon each producing a magnetic field, cause the Hb resonance to be split into a triplet -CHb3- is ‘split’ into a triplet by two adj. Has -+ ++ +- -Resonance, n, in absence of coupling CHEM 430 – NMR Spectroscopy 96 NMR Spectroscopy The Coupling Constant 2-6 • Rather than having to do this exercise for every situation, it is quickly recognized that a given family of equivalent protons (in the absence of other spin-coupling) will have its resonance split into a multiplet containing n+1 peaks, where n is the number of hydrogens on carbons adjacent to the carbon bearing the proton giving the resonance – this is the n + 1 rule # of Hs on adj. C’s Multiplet # of peaks 0 singlet 1 1 1 doublet 2 1 1 2 triplet 3 1 2 1 3 quartet 4 1 3 3 1 4 quintet 5 1 4 6 4 1 5 sextet 6 1 5 10 10 5 1 6 septet 7 1 6 15 20 15 6 1 The relative ratios of the peaks are a mathematical progression given by Pascal’s triangle: CHEM 430 – NMR Spectroscopy 97 NMR Spectroscopy The Coupling Constant 2-6 • Common patterns: tert-butyl - singlet methyl - singlet ethyl – quartet - triplet n-propyl – triplet - quintet - triplet iso-propyl – septet - doublet CHEM 430 – NMR Spectroscopy 98 NMR Spectroscopy 2-6 The Coupling Constant CHEM 430 – NMR Spectroscopy 99 NMR Spectroscopy 2-6 The Coupling Constant CHEM 430 – NMR Spectroscopy 100 NMR Spectroscopy 2-6 The Coupling Constant • Heteronuclear coupling between 1H and 13C are not apparent in 1H spectra (13C low abundance - 1.1%), In 99 of 100 cases, 1H are attached to nonmagnetic 12C atoms. • In the 13C spectrum, carbon nuclei are coupled to 1H directly (99% abundant) attached to the carbon. • Thus, the 13C resonance of a methyl carbon is split into a quartet, that of a methylene carbon CH2 into a triplet, and that of a methine carbon CH into a doublet. A quaternary carbon is not split by one bond coupling. CHEM 430 101 NMR Spectroscopy 2-6 The Coupling Constant • Shown here (top) the 13C spectrum of 3- hydroxybutyric acid which contains a carbon resonance with each type of multiplicity. undecoupled • From right to left are seen a quartet CH3 , a triplet CH2 , a doublet CH , and a singlet CO2H (C=O) • Instrumental procedures, called decoupling, are available by which spin– spin split-tings may be removed. These methods, discussed in Section 5- 3, involve irradiating one CHEM 430 decoupled 102 NMR Spectroscopy 2-6 The Coupling Constant • Instrumental procedures, called decoupling, are available by which spin– spin splittings are removed for clarity. undecoupled • These methods involve irradiating one nucleus with an additional field B2 while observing another nucleus resonating in the B1 field. • 13C spectra are usually run decoupled as other spectral techniques are used to establish 1H-13C connectivity more rapidly and with more clarity CHEM 430 decoupled 103 The NMR Spectrum - 1H NMR Spectroscopy Spin-spin splitting – 1H NMR The next level of complexity (which we will cover in detail in Chapter 4) is when protons on adjacent carbons exert different J’s than one another. Consider the ethylene fragment: The influence of the geminal-relationship is over the shortest distance The magnetic influence of the transrelationship is over the longest distance The cis-relationship, is over an intermediate distance CHEM 430 – NMR Spectroscopy 104 The NMR Spectrum - 1H NMR Spectroscopy Spin-spin splitting – 1H NMR For ethylene we would then observe three chemically distinct resonances with spin-spin splitting exerted by the other two protons: J couplings: 2J gem = 0 – 1 Hz The observed multiplet for Ha is a “doublet of doublets” 3J AC 3J trans = 11- 18 Hz 3J AB 3J cis 3J AB = 6 - 15 Hz CHEM 430 – NMR Spectroscopy 105 The NMR Spectrum - 1H NMR Spectroscopy Spin-spin splitting – 1H NMR Similar behavior is observed with aromatic rings; since the ring structure is fairly rigid and electronic effects are conducted over a longer distance, J – couplings are observed across the ring system: 3J ortho 4J meta 5J para In low-field 1H NMR the signal for this proton would be split into a doublet by the proton ortho to it. On a high field instrument one finds this 3J 4 5 ortho as well as a Jmeta and a J para from the effect of the protons meta and para to it Typically: 3J ortho = 7-10 Hz 4J meta = 1-3 Hz 5J para = 0-1 Hz CHEM 430 – NMR Spectroscopy 106 The NMR Spectrum - 1H NMR Spectroscopy Spin-spin splitting – 1H NMR For our initial treatment of 1H NMR the alkenyl, aromatic and the following J values should be learned: 3J 3J = 6-8 = 8-14 3J a,e = 0-7 3J e,e = 0-5 3J trans = 11-18 3J = 6-15 cis a,a 3J = 8-11 3J = 5-7 3J = 4-8 3J = 6-12 cis trans 3J 3J allyl = 4-10 ortho = 7-10 Hz meta = 1-3 Hz 5J para = 0-1 Hz 4J 3J = 4-8 3J = 6-12 cis CHEM 430 – NMR Spectroscopy trans 107 The NMR Spectrum - 1H A typical 1H NMR is recorded from -2 to 15 d (ppm); what is typically reported is the region from 0 to 10 d Remember, if a proton is shielded (e- circulation reduces “felt” magnetic field) DE for the transition is lowered and the signal is near the high field or upfield region of the spectrum (right) If the proton is deshielded (e- circulation doesn’t reduce the “felt” magnetic field) DE for the transition is raised and the signal is near the low field or downfield region of the spectrum (left) high DE deshielded 1H sees full B0 downfield 10 or ppm CHEM 430 –dNMR Spectroscopy low DE shielded 1H reduces B0 upfield 0 108 The NMR Spectrum - 1H The number of signals observed will be equal to the number of unique populations of chemically equivalent protons To determine if two protons are chemically equivalent, substitute “X” for that each respective hydrogen in the compound and compare the structures If the two structures are fully superimposible (identical) the two hydrogens are chemically equivalent; if the two structures are different the two hydrogens were not chemically equivalent CH3 H A simple example: p-xylene CH3 X H CH3 Same structure H CH3 CHEM 430 – NMR Spectroscopy CH3 X H CH3 109 The NMR Spectrum - 1H The position (v) of each resonance is dependant on the electronic environment around the proton – chemical shift as a result of local diamagnetic shielding There are three principle effects that contribute to local diamagnetic shielding: 1) Electronegativity 2) Hybridization 3) Proton acidity/exchange CHEM 430 – NMR Spectroscopy 110 The NMR Spectrum - 1H Local Diamagnetic Shielding - Electronegativity 1. Electronegative groups comprise most organic functionalities: -F -Cl -Br -I -OH -OR -NH2 -NHR -NR2 -NH3+ -C=O -PO3H2 -SH -Ph -C=C -NO2-NO -SO3H and most others In all cases, the inductive w/d of electrons of these groups decreases the electron density in the C-H covalent bond – proton is deshielded – higher DE of transition CHEM 430 – NMR Spectroscopy 111 The NMR Spectrum - 1H Local Diamagnetic Shielding - Electronegativity 2. Protons bound to carbons bearing electron withdrawing groups are deshielded based on the magnitude of the withdrawing effect – Pauling electronegativity: CH3F CH3O- CH3Cl CH3Br CH3I CH4 (CH3)4Si Pauling Electronegativity 4.0 3.5 3.1 2.8 2.5 2.1 1.8 d of H 4.26 3.40 3.05 2.68 2.16 0.23 0.0 CHEM 430 – NMR Spectroscopy 112 The NMR Spectrum - 1H Local Diamagnetic Shielding - Electronegativity 3. The magnitude of the withdrawing effect is cumulative: CH3Cl CH2Cl2 CHCl3 3.05 5.30 7.27 d of H 4. The magnitude of the withdrawing effect is reduced by distance, as the inductive model suggests d of H -CH2Br -CH2CH2Br -CH2CH2CH2Br 3.30 1.69 1.25 CHEM 430 – NMR Spectroscopy 113 The NMR Spectrum - 1H NMR Spectroscopy Local Diamagnetic Shielding - Hybridization 1. The hybridization of the carbon the proton is bound exerts a strong electronic effect 2. The greater the s-character, the more tightly bound the electrons are to carbon, raising its effective electronegativity (sp = 50% s, sp2, 33% s and sp3 25% s) Type of H Name of H Chemical Shift, d R-CH3, R2CH2, R3CH alkyl 0.8-1.7 C=C-CH3 allyl 1.6-2.6 CC-H Acetylenic 2.0-3.0 C=C-H Vinylic 4.6-5.7 Ar-H aromatic 6.5-8.5 O=C-H aldehydic 9.5-10.1 CHEM 430 – NMR Spectroscopy Something odd is happening here, as we will discuss 114 The NMR Spectrum - 1H NMR Spectroscopy Local Diamagnetic Shielding - Proton Acidity/Exchange 1. If an organic molecule that possesses hydrogen atoms of low pKA are dissolved in a deuterated solvent that also has a low pKA, the “visible” protons will exchange with “deuterium” from solvent and become “invisible” to the NMR spectrometer OH OD D2O Such studies are useful, if it is desired to see which H-atoms on an organic are acidic! CHEM 430 – NMR Spectroscopy 115 The NMR Spectrum - 1H NMR Spectroscopy Local Diamagnetic Shielding - Proton Acidity/Exchange 2. Due to H-bonding effects, the resonance for certain functional groups (esp. –OH and – NH2) can change drastically dependent on concentration and the extent of the H- bonding 3. Just as in IR spectroscopy, peaks corresponding to these resonances are broad and often undefined – observing a continuum of bond strengths/electron densities about the observed proton 4. The correlation tables for the position of such protons tend to be broad and unreliable: • • • • • • Acid –OH Phenol –OH Alcohol –OH Amine –NH2 Amide –NH2 Enol CH=CH-OH 10.5-12.0 d 4.0-12.0 d 0.5-5.0 d 0.5-5.0 d 5.0-8.0 d >15 d CHEM 430 – NMR Spectroscopy 116 NMR Spectroscopy Some observed 1H resonances can not be fully explained by local diamagnetic shielding effects Magnetic Anisotropy – literally “magnetic dissimilarity” For example, by our hybridization model, a proton bound to an sp2 C should be observed at lower d than a proton bound to an sp C Type of H Name of H Chemical Shift, d R-CH3, R2CH2, R3CH alkyl 0.8-1.7 C=C-CH3 allyl 1.6-2.6 CC-H Acetylenic 2.0-3.0 C=C-H Vinylic 4.6-5.7 Ar-H aromatic 6.5-8.5 O=C-H aldehydic 9.5-10.1 CHEM 430 – NMR Spectroscopy 117 The NMR Spectrum - 1H NMR Spectroscopy Magnetic Anisotropy – 1. This effect is primary due to the fact that there is an additional effect of circulating electrons, observed in p-systems 2. In benzene, the 6-p-orbitals overlap to allow full circulation of electrons; as these electrons circulate in the applied magnetic field they oppose the applied magnetic field at the center – just like the circulation of electrons in the 1-s orbital about hydrogen – at the middle!: B0 CHEM 430 – NMR Spectroscopy 118 The NMR Spectrum - 1H NMR Spectroscopy Magnetic Anisotropy – 3. On the periphery of the ring, the effect is opposite – the magnetic effect reinforces the applied B0, and DE becomes greater – deshielding effect CHEM 430 – NMR Spectroscopy 119 The NMR Spectrum - 1H NMR Spectroscopy Magnetic Anisotropy – 4. This theory can easily be tested by the observation of large aromatic systems that possess protons inside the ring (now a shielding effect): -1.8 d 8.9 d Or over a ring system: H H 2.0 d H2 C CH2 CHEM 430 – NMR Spectroscopy -1.0 d 120 The NMR Spectrum - 1H NMR Spectroscopy Magnetic Anisotropy – 5. In alkynes, a similar situation (to the central protons in large aromatic systems) arises where the terminal proton is in the region of maximum shielding CHEM 430 – NMR Spectroscopy 121 The NMR Spectrum - 1H NMR Spectroscopy General Correlation Chart – 1H NMR Due to the three effects on local diamagnetic shielding, in conjunction with the effect of magnetic anisotropy 1H NMR chemical shifts are variable • Avoid using hard and fast rules (tables of numbers) • Instead, start from the general correlation table and deduce structural features based on the effects just discussed • After a structural inference has been made, then use the more specific correlation tables to confirm the analysis CHEM 430 – NMR Spectroscopy 122 The NMR Spectrum - 1H NMR Spectroscopy General Correlation Chart – 1H NMR CH3 Here are the general regions for 1H chemical shifts: H Si O H3C C H C CH CH3 3 O C H O H S H R O H O O H O H C C Ph 15 10 9 8 7 downfield deshielded higher DE 6 R R H C C C 5 4 d (ppm) CHEM 430 – NMR Spectroscopy R = H or alkyl EWG 3 2 1 0.0 upfield shielded lower DE 123 The NMR Spectrum - 1H NMR Spectroscopy Spin-spin splitting – 1H NMR 1. The magnetic effects of nuclei in close proximity to those being observed have an effect on the local magnetic field, and therefore DE 2. Specifically, when proton is close enough to another proton, typically by being on an adjacent carbon (vicinal), it can “feel” the magnetic effects generated by that proton 3. On any one of the 108 of these molecules in a typical NMR sample, there is an equal statistical probability that the adjacent (vicinal) proton is either in the + ½ or – ½ spin state 4. If there is more than one proton on an adjacent carbon – all the statistical probabilities exist that each one is either + ½ or – ½ in spin 5. The summation of these effects over all of the observed nuclei in the sample is observed as the spin-spin splitting of resonances CHEM 430 – NMR Spectroscopy 124 Intensity of Signals—Integration • The area under an NMR signal is proportional to the number of absorbing protons • An NMR spectrometer automatically integrates the area under the peaks, and prints out a stepped curve (integral) on the spectrum • The height of each step is proportional to the area under the peak, which in turn is proportional to the number of absorbing protons • Modern NMR spectrometers automatically calculate and plot the value of each integral in arbitrary units • The ratio of integrals to one another gives the ratio of absorbing protons in a spectrum; note that this gives a ratio, and not the absolute number, of absorbing protons CHEM 430 – NMR Spectroscopy 125 The NMR Spectrum - 1H NMR Spectroscopy Integration – 1H NMR 1. Like instrumental chromatography, in NMR spectroscopy, the area under a peak (or multiplet) is proportional to the number of protons in the sample that generated that particular resonance 2. The NMR spectrometer typically will print this information on the spectrum as an integral line (stepped line on the spectrum below) 3. The height of the integral is proportional to that proton population; by comparing the ratios of the integrals on an NMR spectrum you can determine the number of protons as a least common multiple of these ratios CHEM 430 – NMR Spectroscopy 126 The NMR Spectrum - 1H NMR Spectroscopy Integration – 1H NMR 4. For example observe the integration of the ethanol spectrum below: HO CH3 C H2 -CH3 3.75 units high -OH 1.25 units high 2.5 units high CHEM 430 – NMR Spectroscopy 127 Intensity of Signals—Integration CHEM 430 – NMR Spectroscopy 128 Intensity of Signals—Integration CHEM 430 – NMR Spectroscopy 129 Nuclear Magnetic Resonance When a nuclei of spin +½ encounters a photon where n = E/h, the two “couple” The nuclei “tips” its spin state and is now opposed to B0 CHEM 430 – NMR Spectroscopy The nuclei “relaxes” and returns to the + ½ spin state 130 Basis of NMR Spectroscopy 2.1 Excellent Simulators for NMR Phenomenon • 2-D compass needle analogy: http://www.drcmr.dk/JavaCompass/ • 3-D Bloch equation simulator http://www.drcmr.dk/BlochSimulator/ CHEM 430 – NMR Spectroscopy 131 General Theory NMR Spectroscopy Nuclear Spin States • The sub-atomic particles within atomic nuclei possess a spin quantum number just like electrons • As with electrons, the nucleons are organized in energy levels • Just as when using Hund’s rules to fill atomic orbitals with electrons, nucleons must each have a unique set of quantum numbers • The total spin quantum number of a nucleus is a physical constant, I • For each nucleus, the total number of spin states allowed is given by the equation: 2I + 1 CHEM 430 – NMR Spectroscopy 132 General Theory NMR Spectroscopy Nuclear Spin States Spin Quantum Numbers of Common Nuclei Element 1H 2H 12C 13C 14N 16O 17O 19F 31P 35Cl Nuclear Spin Quantum Number ½ 1 0 ½ 1 0 5/2 ½ ½ 3/2 # of spin states 2 3 0 2 3 0 6 2 2 4 6. Observe that for atoms with no net nuclear spin, there are zero allowed spin states 7. All the spin states of a given nucleus are degenerate in energy CHEM 430 – NMR Spectroscopy 133 General Theory NMR Spectroscopy Nuclear Magnetic Moments • A nucleus contains protons, which each bear a +1 charge • If the nucleus has a net nuclear spin, and an odd number of protons, the rotation of the nucleus will generate a magnetic field along the axis of rotation • Thus, a nucleus has a magnetic moment, m, generated by its charge and spin • A hydrogen atom with its lone proton making up the nucleus, can have two possible spin states, degenerate in energy m +½ H m H CHEM 430 – NMR Spectroscopy -½ 134 General Theory Nuclear Spin States 5. In the presence of an externally applied magnetic field, these two spin states are no longer degenerate in energy 6. The spin opposed orientation is slightly higher in energy than the spin aligned orientation m -½ H B0 – externally applied magnetic field DE m +½ H CHEM 430 – NMR Spectroscopy 135 General Theory Absorption of Energy • The energy difference between the two non-degenerate spin states in the presence of an applied magnetic field is quantized • At low B0 is easy to surmise that the potential energy of the spin opposed state would be low, and as B0 grows in strength, so would the potential energy • Thus, with increasing strength of B0, DE between the two spin states also increases -1/2 -1/2 +1/2 DE +1/2 CHEM 430 –BNMR Spectroscopy 0 – increasing 136 General Theory NMR Spectroscopy Absorption of Energy 4. From theory we have already discussed, we say that a quantum mechanical particle can absorb a photon of energy equal to DE and become promoted to the higher state 5. This energy is proportional to the frequency of the photon absorbed, and in the case of nuclear spin, is a function of the magnetic field applied: DE = hn = f (B0) 6. Every nucleus has a different ratio of m to angular momentum (each has a different charge and mass) – this is referred to as the magnetogyric ratio, g DE = hn = f (gB0) Angular momentum is quantized in units of h/2p, thus: DE = hn = g (h/2p)B CHEM 0 430 – NMR Spectroscopy 137 General Theory Absorption of Energy 7. Solving for the frequency of EM radiation we are observing: DE = n = (g/2p) B0 8. For a bare hydrogen nucleus (H+), g = 267.53 (106 radians/T·sec) 9. In a field strength of 1 Tesla, DE = 42.5 MHz (for our discussion, at 1.41 T, DE = 60 MHz) DE = hn = f (B0) This energy difference corresponds to the highly weak radio frequency region of the EM spectrum – with wavelengths of >5 meters equal to < 0.02 cal·mol-1 g-rays X-rays UV IR Microwave Radio CHEM 430 – NMR Spectroscopy 138 General Theory Mechanism of absorption – nuclear magnetic resonance • What we are actually observing for DE is the precessional or Larmor frequency (w) of the spinning nucleus – this is analogous to a spinning toy top precessing as a result of the influence of the earth’s magnetic field: w B0 CHEM 430 – NMR Spectroscopy 139 General Theory NMR Spectroscopy Mechanism of absorption – Nuclear Magnetic Resonance 2. When a photon of n = 60 MHz encounters this spinning charged system (a bare proton) the two can couple and change the spin state of the proton This state is called nuclear magnetic resonance, and the nucleus is said to be in resonance with the incoming radio wave w DE B0 n=w w CHEM 430 – NMR Spectroscopy 140 General Theory NMR Spectroscopy Mechanism of absorption – Nuclear Magnetic Resonance 3. The energy difference corresponding to 60 MHz (DE = hn) is 2.39 x 10-5 kJ mol-1 (tiny) – thermal energy at room temperature (298 oK) is sufficient to populate both energy levels 4. The energy difference is small, so rapid exchange is occurring between the two populations, but there is always a net excess of protons in the lower energy state 5. From the Boltzman distribution equation we can calculate the population of each energy state: Nupper/Nlower = e-DE/kT = e-hn/kT @ 298 oK the ratio is 1,000,000 / 1,000,009 ! There is an excess population of 9 nuclei in the lower energy state! CHEM 430 – NMR Spectroscopy 141 General Theory Mechanism of absorption – Nuclear Magnetic Resonance 6. As the applied B0 increases, exchange becomes more difficult and the excess increases: Frequency (MHz) Excess nuclei 60 9 80 12 100 16 200 32 300 48 600 96 7. In each case, it is these few nuclei that allow us to observe NMR 8. When radio radiation is applied to a sample both transitions upward and downward are stimulated – if too much radiation is applied both states completely equilibrate – a state called saturation – no NMR signal can be observed CHEM 430 – NMR Spectroscopy 142 General Theory NMR Spectroscopy Chemical Shift 6. Spectroscopic observation of the NMR phenomenon would be of little use if all protons resonated at the same frequency 7. The protons in organic compounds are not bare nuclei, they are surrounded by an s -orbital of containing an electron shared with an electron in a hybridized orbital of another atom to form a covalent bond 8. In the presence of an external magnetic field, an induced circulation of electrons opposite to that of a proton is observed since the two are of opposite charges 9. This induced circulation generates a magnetic field in opposition to the applied magnetic field – a local diamagnetic current CHEM 430 – NMR Spectroscopy 143 Basis of NMR Spectroscopy 2.1 Magnetic Properties of Nuclei • The magnitude of m from atom to atom varies: m = għI • ħ is Planck’s constant divided by 2p • g is the characteristic gyromagnetic ratio of the nucleus • The larger g is, the greater the magnetic moment CHEM 430 – NMR Spectroscopy 144 Nuclear Magnetic Resonance • For the 1H nucleus (proton) this resonance condition occurs at low energy (lots of noise) unless a very large magnetic field is applied • Early NMR spectrometers used a large permanent magnet with a field of 1.4 Tesla—protons undergo resonance at 60 MHz (1 MHz = 106 Hz) • Modern instruments use a large superconducting magnet—our NMR operates at 9.4 T where proton resonance occurs at 400 MHz • In short, higher field gives cleaner spectra and allows longer and more detailed experiments to be performed CHEM 430 – NMR Spectroscopy 145 Origin of the Chemical Shift Electrons surrounding the nucleus are opposite in charge to the proton, therefore they generate an opposing b0 Deshieding Shielding Factors which lower edensity allow the nucleus to “see” more of the B0 being applied – resonance occurs at higher energy Factors which raise e- density reduce the amount of B0 the nucleus “sees” – resonance condition occurs at lower energy CHEM 430 – NMR Spectroscopy 146 The Proton (1H) NMR Spectrum CHEM 430 – NMR Spectroscopy 147 The 1H NMR Spectrum A reference compound is needed—one that is inert and does not interfere with other resonances Chemists chose a compound with a large number of highly shielded protons—tetramethylsilane (TMS) No matter what spectrometer is used the resonance for the protons on this compound is set to d 0.00 CH3 Si H3C CH CH3 3 CHEM 430 – NMR Spectroscopy 148 The 1H NMR Spectrum The chemical shift for a given proton is in frequency units (Hz) This value will change depending on the B0 of the particular spectrometer By reporting the NMR absorption as a fraction of the NMR operating frequency, we get units, ppm, that are independent of the spectrometer CHEM 430 – NMR Spectroscopy 149 The 1H NMR Spectrum • We need to consider four aspects of a 1H spectrum: a. b. c. d. Number of signals Position of signals Intensity of signals. Spin-spin splitting of signals CHEM 430 – NMR Spectroscopy 150 The Number of Signals The number of NMR signals equals the number of different types of protons in a compound Protons in different environments give different NMR signals Equivalent protons give the same NMR signal CHEM 430 – NMR Spectroscopy 151 The Number of Signals To determine if two protons are chemically equivalent, substitute “X” for that each respective hydrogen in the compound and compare the structures If the two structures are fully superimposible (identical) the two hydrogens are chemically equivalent; if the two structures are different the two hydrogens were not equivalent CH 3 A simple example: p-xylene H CH 3 Z H CH 3 Same Compound H CH 3 CH 3 Z CHEM 430 – NMR Spectroscopy H CH 3 152 The Number of Signals • Examples Important: To determine equivalent protons in cycloalkanes and alkenes, always draw all bonds to show specific stereochemistry: CHEM 430 – NMR Spectroscopy 153 The Number of Signals In comparing two H atoms on a ring or double bond, two protons are equivalent only if they are cis or trans to the same groups. CHEM 430 – NMR Spectroscopy 154 The Number of Signals Proton equivalency in cycloalkanes can be determined similarly: CHEM 430 – NMR Spectroscopy 155 The Number of Signals Enantiotopic Protons – when substitution of two H atoms by Z forms enantiomers: a. The two H atoms are equivalent and give the same NMR signal b. These two atoms are called enantiotopic CHEM 430 – NMR Spectroscopy 156 The Number of Signals Diastereotopic Protons - when substitution of two H atoms by Z forms diastereomers a. The two H atoms are not equivalent and give two NMR signals b. These two atoms are called diastereotopic CHEM 430 – NMR Spectroscopy 157 Chemical Shift – Position of Signals • Remember: Electrons surrounding the nucleus are opposite in charge to the proton, therefore they generate an opposing b0 Deshieding Shielding Factors which lower e- density allow the nucleus to “see” more of the B0 being applied – resonance occurs at higher energy Factors which raise e- density reduce the amount of B0 the nucleus “sees” – resonance condition occurs at lower energy CHEM 430 – NMR Spectroscopy 158 Chemical Shift – Position of Signals • The less shielded the nucleus becomes, the more of the applied magnetic field (B0) it feels • This deshielded nucleus experiences a higher magnetic field strength, to it needs a higher frequency to achieve resonance • Higher frequency is to the left in an NMR spectrum, toward higher chemical shift—so deshielding shifts an absorption downfield Downfield, deshielded CHEM 430 – NMR Spectroscopy Upfield, shielded 159 Chemical Shift – Position of Signals • There are three principle effects that contribute to local diamagnetic shielding: a. Electronegativity b. Hybridization c. Proton acidity/exchange CHEM 430 – NMR Spectroscopy 160 Chemical Shift – Position of Signals Electronegative groups comprise most organic functionalities: -F -Cl -Br -I -NHR -NR2 -NH3+ -PO3H2 -SH -Ph -C=C -OH -OR -NH2 -C=O -NO2-NO -SO3H and most others In all cases, the inductive WD of electrons of these groups decreases the electron density in the C-H covalent bond – proton is deshielded – signal more downfield of TMS CHEM 430 – NMR Spectroscopy 161 Chemical Shift – Position of Signals Protons bound to carbons bearing electron withdrawing groups are deshielded based on the magnitude of the withdrawing effect – Pauling electronegativity: CH3F CH3O- CH3Cl CH3Br CH3I CH4 (CH3)4Si Pauling Electronegativity 4.0 3.5 3.1 2.8 2.5 2.1 1.8 d of H 4.26 3.40 3.05 2.68 2.16 0.23 0.0 CHEM 430 – NMR Spectroscopy 162 Chemical Shift – Position of Signals 3. The magnitude of the deshielding effect is cumulative: CH3Cl CH2Cl2 CHCl3 3.05 5.30 7.27 d of H As more chlorines are added d becomes larger 3. The magnitude of the deshielding effect is reduced by distance, as the inductive model suggests d of H -CH2Br -CH2CH2Br -CH2CH2CH2Br 3.30 1.69 1.25 CHEM 430 – NMR Spectroscopy 163 Chemical Shift – Position of Signals Hybridization Increasing s-character (sp3 sp2 sp) pulls e- density closer to nucleus effectively raising electronegativity of the carbon the H atoms are bound to – a deshielding effect We would assume that H atoms on sp carbons should be well downfield (high d) and those on sp3 carbons should be upfield (low d) CHEM 430 – NMR Spectroscopy 164 Chemical Shift – Position of Signals • What we observe is slightly different: Type of H Carbon hybridization Name of H Chemical Shift, d R-CH3, R2CH2, R3CH sp3 alkyl 0.8-1.7 C=C-CH3 sp3 allyl 1.6-2.6 CC-H sp acetylenic 2.0-3.0 C=C-H sp2 vinylic 4.6-5.7 Ar-H sp2 aromatic 6.5-8.5 O=C-H sp2 aldehydic 9.5-10.1 Chemists refer to this observation as magnetic anisotropy CHEM 430 – NMR Spectroscopy 165 Chemical Shift – Position of Signals Magnetic Anisotropy – Aromatic Protons a. In a magnetic field, the six p electrons in benzene circulate around b. c. the ring creating a ring current. The magnetic field induced by these moving electrons reinforces the applied magnetic field in the vicinity of the protons. The protons thus feel a stronger magnetic field and a higher frequency is needed for resonance. Thus they are deshielded and absorb downfield. CHEM 430 – NMR Spectroscopy 166 Chemical Shift – Position of Signals • Similarly this effect operates in alkenes: CHEM 430 – NMR Spectroscopy 167 Chemical Shift – Position of Signals • In alkynes there are two perpendicular sets of p-electrons—the molecule orients with the field lengthwise—opposing B0 shielding the terminal H atom CHEM 430 – NMR Spectroscopy 168 Chemical Shift – Position of Signals CHEM 430 – NMR Spectroscopy 169 Chemical Shift – Position of Signals CHEM 430 – NMR Spectroscopy 170 Intensity of Signals—Integration The area under an NMR signal is proportional to the number of absorbing protons An NMR spectrometer automatically integrates the area under the peaks, and prints out a stepped curve (integral) on the spectrum The height of each step is proportional to the area under the peak, which in turn is proportional to the number of absorbing protons Modern NMR spectrometers automatically calculate and plot the value of each integral in arbitrary units The ratio of integrals to one another gives the ratio of absorbing protons in a spectrum; note that this gives a ratio, and not the absolute number, of absorbing protons CHEM 430 – NMR Spectroscopy 171 Intensity of Signals—Integration CHEM 430 – NMR Spectroscopy 172 Intensity of Signals—Integration CHEM 430 – NMR Spectroscopy 173 Spin-Spin Splitting • Consider the spectrum of ethyl alcohol: • Why does each resonance “split” into smaller peaks? HO CH3 C H2 CHEM 430 – NMR Spectroscopy 174 Spin-Spin Splitting The magnetic effects of nuclei in close proximity to those being observed have an effect on the local magnetic field, and therefore DE Specifically, when proton is close enough to another proton, typically by being on an adjacent carbon (vicinal), it can “feel” the magnetic effects generated by that proton On any one of the 108 of these molecules in a typical NMR sample, there is an equal statistical probability that the adjacent (vicinal) proton is either in the + ½ or – ½ spin state If there is more than one proton on an adjacent carbon – all the statistical probabilities exist that each one is either + ½ or – ½ in spin The summation of these effects over all of the observed nuclei in the sample is observed as the spin-spin splitting of resonances CHEM 430 – NMR Spectroscopy 175 Spin-Spin Splitting • Recall, we are observing the frequency (E = hn) where a proton goes into resonance Any change in B0 will cause a change in energy at which the resonance condition will occur for a proton of a given chemical shift CHEM 430 – NMR Spectroscopy 176 In solution we are not looking at a single molecule but about 108 On some molecules the proton being observed may be next to another proton of spin + 1/2 : CHEM 430 – NMR Spectroscopy 177 Spin-Spin Splitting • On some molecules the proton being observed may be next to another proton of spin – 1/2 : CHEM 430 – NMR Spectroscopy 178 Spin-Spin Splitting Observe what effect this has on an isolated ethyl group: The two methylene Ha protons have three neighbors, Hb, on the adjacent methyl carbon Each one of these hydrogens can be + ½ or – ½ , and since we are not looking at one molecule, but billions, we will observe all combinations CHEM 430 – NMR Spectroscopy 179 Spin-Spin Splitting The first possibility is that all three Hb protons have a + ½ spin; in this case the three protons combine to generate three small magnetic fields that aid B0 and deshield the protons – pushing the resonance for Ha slightly downfield (the magnetic field of a proton is tiny compared to B0) All 3 Hb protons + ½ CHEM 430 – NMR Spectroscopy resonance for Ha in absence of spin-spin splitting 180 Spin-Spin Splitting The second possibility is that two Hb protons have a + ½ spin and the third a - ½ ; in this case the two protons combine to enhance B0 and the other against it, a net deshielding; there are 3 different combinations that generate this state or or 2 Hb protons + ½ CHEM 430 – NMR Spectroscopy resonance for Ha in absence of spin-spin splitting 181 Spin-Spin Splitting The third possibility is that two Hb protons have a –½ spin and the third +½; here, the two protons combine to reduce B0 and the other enforce it, a net shielding effect; there are 3 different combinations that generate this state or or 2 Hb protons - ½ CHEM 430 – NMR Spectroscopy resonance for Ha in absence of spin-spin splitting 182 Spin-Spin Splitting The last possibility is that all three Hb protons have a – ½ spin; in this case the three protons combine to oppose B0, a net shielding effect; there is one combination that generates this state All 3 Hb protons - ½ CHEM 430 – NMR Spectroscopy resonance for Ha in absence of spin-spin splitting 183 Spin-Spin Splitting The result is instead of one resonance (peak) for Ha, the peak is “split” into four, a quartet, with the constituent peaks having a ratio of 1:3:3:1 centered at the d (n) for the resonance CHEM 430 – NMR Spectroscopy resonance for Ha in absence of spin-spin splitting 184 Spin-Spin Splitting Similarly, the Hb protons having two protons, on the adjacent carbon each producing a magnetic field, cause the Hb resonance to be split into a triplet CHEM 430 – NMR Spectroscopy resonance for Ha in absence of spin-spin splitting 185 Spin-Spin Splitting Rather than having to do this exercise for every situation, it is quickly recognized that a given family of equivalent protons (in the absence of other spin-coupling) will have its resonance split into a multiplet containing n+1 peaks, where n is the number of hydrogens on carbons adjacent to the carbon bearing the proton giving the resonance – this is the n + 1 rule # of Hs on adj. C’s Multiplet # of peaks 0 singlet 1 1 1 doublet 2 1 1 2 triplet 3 1 2 1 3 quartet 4 1 3 3 1 4 quintet 5 1 4 6 4 1 5 sextet 6 1 5 10 10 5 1 6 septet 7 CHEM 430 – NMR Spectroscopy The relative ratios of the peaks are a mathematical progression given by Pascal’s triangle: 1 6 15 20 15 6 1 186 1H NMR—Spin-Spin Splitting • Common patterns: tert-butyl - singlet methyl - singlet ethyl – quartet - triplet n-propyl – triplet - quintet - triplet iso-propyl – septet - doublet CHEM 430 – NMR Spectroscopy 187 1H NMR—Spin-Spin Splitting CHEM 430 – NMR Spectroscopy 188 1H NMR—Spin-Spin Splitting Another Example: Br Br Br = Br C Br Ha C Br Hb Hb CHEM 430 – NMR Spectroscopy 189 1H NMR—Spin-Spin Splitting Another Example: CHEM 430 – NMR Spectroscopy 190 1H NMR—Spin-Spin Splitting Three general rules describe the splitting patterns commonly seen in the 1H NMR spectra of organic compounds: 1.Equivalent protons do not split each other’s signals 2.A set of n nonequivalent protons splits the signal of a nearby proton into n + 1 peaks 3.Splitting is observed for nonequivalent protons on the same carbon or adjacent carbons If Ha and Hb are not equivalent, splitting is observed when: CHEM 430 – NMR Spectroscopy 191 1H NMR—Spin-Spin Splitting • Magnetic influence falls off dramatically with distance • The n + 1 rule only works in the following situations: H H Aliphatic compounds that have free rotation about each bond H H G H Ha H Hb Hc Aromatic compounds where each proton is held in position relative to one another CHEM 430 – NMR Spectroscopy 192 1H NMR—Spin-Spin Splitting The amount of influence exerted by a proton on an adjacent carbon is observed as the difference (in Hz) between component peaks within the multiplet it generates. This influence is quantified as the coupling constant, J Two sets of protons that split one another are said to be “coupled” J for two sets of protons that are coupled are equivalent— therefore on complex spectra we can tell what is next to what This J Is equal to this J -CH2- CHEM 430 – NMR Spectroscopy -CH3 193 1H NMR—Spin-Spin Splitting The next level of complexity (which at this level, is only introduced) is when protons on adjacent carbons exert different J’s than one another. Consider the ethylene fragment: The influence of the geminal-relationship is over the shortest distance The magnetic influence of the transrelationship is over the longest distance The cis-relationship, is over an intermediate distance CHEM 430 – NMR Spectroscopy 194 1H NMR—Spin-Spin Splitting • For this substituted ethylene we see the following spectrum: 2J gem = 0 – 1 Hz The observed multiplet for Ha is a “doublet of doublets” 3J AC 3J trans = 11- 18 Hz 3J AB 3J cis 3J AB = 6 - 15 Hz CHEM 430 – NMR Spectroscopy 195 1H NMR—Spin-Spin Splitting In general, when two sets of adjacent protons are different from each other (n protons on one adjacent carbon and m protons on the other), the number of peaks in an NMR signal = (n + 1)(M + 1) In general the value of J falls off with distance; J values have been tabulated for virtually all alkene, aromatic and aliphatic ring systems CHEM 430 – NMR Spectroscopy 196 1H NMR—Spin-Spin Splitting • Some common J-values 3J 3J = 6-8 = 8-14 3J a,e = 0-7 3J e,e = 0-5 3J trans = 11-18 3J = 6-15 cis a,a 3J allyl = 4-10 3J = 8-11 3J = 5-7 3J = 4-8 3J = 6-12 cis CHEM 430 – NMR Spectroscopy trans 3J = 4-8 3J = 6-12 cis trans 3J ortho = 7-10 Hz 4J meta = 1-3 Hz 5J para = 0-1 Hz 197 1H NMR—Spin-Spin Splitting • We can now tell stereoisomers apart through 1H NMR: CHEM 430 – NMR Spectroscopy 198 1H NMR—Spin-Spin Splitting • A combined example: CHEM 430 – NMR Spectroscopy 199 1H NMR—Spin-Spin Splitting • Under usual conditions, an OH proton does not split the NMR signal of adjacent protons • Protons on electronegative atoms rapidly exchange between molecules in the presence of trace amounts of acid or base (usually with NH and OH protons) CHEM 430 – NMR Spectroscopy 200 Structure Determination CHEM 430 – NMR Spectroscopy 201 Structure Determination CHEM 430 – NMR Spectroscopy 202 Structure Determination CHEM 430 – NMR Spectroscopy 203 Structure Determination CHEM 430 – NMR Spectroscopy 204 13C NMR • The lack of splitting in a 13C spectrum is a consequence of the low natural abundance of 13C • Recall that splitting occurs when two NMR active nuclei—like two protons—are close to each other. Because of the low natural abundance of 13C nuclei (1.1%), the chance of two 13C nuclei being bonded to each other is very small (0.01%), and so no carboncarbon splitting is observed • A 13C NMR signal can also be split by nearby protons. This 1H-13C splitting is usually eliminated from the spectrum by using an instrumental technique that decouples the proton-carbon interactions, so that every peak in a 13C NMR spectrum appears as a singlet • The two features of a 13C NMR spectrum that provide the most structural information are the number of signals observed and the chemical shifts of those signals CHEM 430 – NMR Spectroscopy 205 13C NMR CHEM 430 – NMR Spectroscopy 206 13C NMR • The number of signals in a 13C spectrum gives the number of different types of carbon atoms in a molecule. • Because 13C NMR signals are not split, the number of signals equals the number of lines in the 13C spectrum. • In contrast to the 1H NMR situation, peak intensity is not proportional to the number of absorbing carbons, so 13C NMR signals are not integrated. CHEM 430 – NMR Spectroscopy 207 13C NMR • In contrast to the small range of chemical shifts in 1H NMR (1-10 ppm usually), 13C NMR absorptions occur over a much broader range (0-220 ppm). • The chemical shifts of carbon atoms in 13C NMR depend on the same effects as the chemical shifts of protons in 1H NMR. CHEM 430 – NMR Spectroscopy 208 13C NMR CHEM 430 – NMR Spectroscopy 209 13C NMR CHEM 430 – NMR Spectroscopy 210 Shoolery Tables • After years of collective observation of 1H and 13C NMR it is possible to predict chemical shift to a fair precision using Shoolery Tables • These tables use a base value for 1H and 13C chemical shift to which are added adjustment increments for each group on the carbon atom H X C H H methyl H X C Y H methylene CHEM 430 – NMR Spectroscopy H X C Z Y methine 211 Shoolery Values for Methylene X or Y Substituent Constant X or Y Substituent Constant -H 0.34 -OC(=O)OR 3.01 -CH3 0.68 -OC(=O)Ph 3.27 -C—C 1.32 -C(=O)R 1.50 -CC- 1.44 -C(=O)Ph 1.90 -Ph 1.83 -C(=O)OR 1.46 -CF2- 1.12 -C(=O)NR2 or H2 1.47 -CF3 1.14 -CN 1.59 -F 3.30 -NR2 or H2 1.57 -Cl 2.53 -NHPh 2.04 -Br 2.33 -NHC(=O)R 2.27 -I 2.19 -OH 2.56 CHEM 430 – NMR Spectroscopy -N 3 1.97 -NO2 3.36 212 Shoolery Values for Methine X ,Y or Z Substituent Constant X, Y or Z Substituent Constant -F 1.59 -OC(=O)OR 0.47 -Cl 1.56 -C(=O)R 0.47 -Br 1.53 -C(=O)Ph 1.22 -NO2 1.84 -CN 0.66 -NR2 or H2 0.64 -C(=O)NH2 0.60 -NH3+ 1.34 -SR or H 0.61 NHC(=O)R 1.80 -OSO2R 0.94 -OH 1.14 -CC- 0.79 -OR 1.14 -C=C 0.46 -C(=O)OR 2.07 -OPh 1.79 -Ph CHEM 430 – NMR Spectroscopy 0.99 213 Shoolery Tables • For methyl—use methylene formula and table using the –H value • For methylene—use a base value of 0.23 and add the two substituent constants for X and Y In 92% of cases experimental is within 0.2 ppm • For methine—use a base value of 2.50 and add the three substituent constants for X, Y and Z Error similar to methylene CHEM 430 – NMR Spectroscopy 214 Shoolery Tables • Work for aromatics as well (.pdf posted) CHEM 430 – NMR Spectroscopy 215 Running an NMR Experiment • Sample sizes for a typical high-field NMR (300-600 MHz): • 1-10 mg for 1H NMR • 10-50 mg for 13C NMR • Solution phase NMR experiments are much simpler to run; solid- phase NMR requires considerable effort • Sample is dissolved in ~1 mL of a solvent that has no 1H hydrogens • Otherwise the spectrum would be 99.5% of solvent, 0.5% sample! CHEM 430 – NMR Spectroscopy 216 Running an NMR Experiment • Deuterated solvents are employed—all 1H atoms replaced with 2H which resonates at a different frequency • Most common: CDCl3 and D2O • Employed if necessary: CD2Cl2, DMSO-d6, toluene-d8, benzene-d6, CD3OD, acetone-d6 • Sample is contained in a high-tolerance thin glass tube (5 mm) CHEM 430 – NMR Spectroscopy 217 Running an NMR Experiment • IMPORTANT—no deuterated solvent is 100% deuterated, there is always residual 1H material, and this will show up on the spectrum • CHCl3 in CDCl3 is a singlet at d 7.27 • HOD in D2O is a broad singlet at d 4.8 • No attempt is made to make solvents for 13C NMR free of 13C, as the resonances are so weak to begin with • NMR using CDCl3 shows a unique 1:1:1 triplet at d 77.00 (+1, 0, 1 spin states of deuterium coupled with 13C) 13C CHEM 430 – NMR Spectroscopy 218