August 31, 2012 - Dipika Aggarwal
advertisement

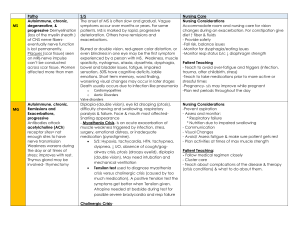
Dipika Aggarwal PGY 4 Neurology Teen aged male admitted with acute onset generalized weakness for 1 day duration Woke up with diffuse weakness; no anti gravity strength in arms, unable to get out of bed Proximal > distal weakness; bilaterally symmetrical Denied diplopia, dysphagia, dysarthria, facial droop, drooling or change in level of consciousness 2 Denied any sensory complains Denied trouble breathing, urinary or bowel incontinence Denied any recent illness, trauma, travel or tick bite One episode of non-bloody emesis prior to admission 3 PMH: similar episode four months earlier, admitted to OSH for 4-5 days, ?? Diagnosed with GBS, ?? treated with plasmapheresis, no LP/ EMG PSH: none Home meds: None FH: HTN, migraine, DM, asthma, no similar problem in family members SH: denies smoking, ETOH or illicit drug use 4 Vitals stable General physical exam unremarkable Neurological exam ◦ Mental status: AAO * 4 ◦ Speech : fluent with comprehension intact ◦ CN 2-12: PERRLA, EOMI, normal facial sensation and symmetry, normal facial strength, hearing intact, equal palatal elevation and tongue midline 5 ◦ Motor: Flaccid tone, motor strength 2/5 proximally and 3-4/5 distally BUE and BLE ◦ DTRs: Areflexic , toes downgoing ◦ Sensory: Intact to LT/PP/ Vibration and proprioception ◦ Unable to test for cerebellar function and gait 6 Where?? What?? 7 Hb - 14.6, WBC 6.1, Plt count 215 Sodium 143, K 1.3, Chloride 110, BUN 13, Creatinine 0.83, Glucose 151, Calcium 9.3, Magnesium 2.0, Phosphorus 2.4 CK 493, Aldolase 15.7 (on day 3) TSH: 2.082, free T3 – 3.8, free T4 – 0.9 Urine lytes: unremarkable 8 Aggressive Potassium replacement Started showing improvement in muscle strength on day 1 By day 2 – strength was 5/5 BUE and BLE Diagnosed with familial hypokalemic periodic paralysis Discharged with follow up care with Jayhawk clinic 9 Non dystrophic myotonias ◦ Myotonia congenita (CLCN1) ◦ Paramyotonia congenita (SCN4A) ◦ Sodium channel myotonias (potassium aggravated myotonias) (SCN4A) Periodic paralyses ◦ Hypokalemic (CACNA1S/ SCN4A) ◦ Hyperkalemic (SCN4A) ◦ Anderson Tawil syndrome (KCNJ2) 10 Hypokalemic: ◦ Thyrotoxic periodic paralysis ◦ hyperaldosteronism ◦ RTA ◦ villous adenoma ◦ cocaine binge ◦ diuretics, licorice, steroids, ETOH Hyperkalemic (k>7): ◦ hyporenemic hypoaldosteronism (DM/CRF) ◦ oral K, CRF, chronic heparin, rhabdomyolysis Normakalemic: ◦ Guanidine, sleep paralysis, MG, TIA, conversion 11 HypoKPP1 and 2 - CACNA1S/ SCN4A gene HypoKPP 1 is the most frequent form 1 in 100,000 Autosomal dominant inheritance pattern M:F – 3 or 4:1 Onset: first 2 decades of life 12 Flaccid paralysis – mild focal weakness to severe generalized weakness Occur anytime of the day; more common in morning Absence of myotonia Proximal > distal weakness; legs > arms Sparing of facial, ventilatory and sphincter muscles Lasts several hours to more than a day 13 Frequency: highly variable Frequency decreases after age 30; may become attack free in 40s and 50s Permanent fixed weakness or slowly progressive weakness more common with HypoKPP1 Attacks may be preceded by sensation of heaviness and or aching in the low back 14 Strenuous physical activity followed by rest or sleep High carb diet ETOH consumption Emotional stress Concurrent viral illness Lack of sleep Medications like beta agonists, corticosteroids, and insulin 15 Mutations in voltage sensor segment D2S4 of 1 subunit of skeletal muscle Ca channel gene, chromosome 1q Arg528His, Arg1239His, Arg1239Gly Less commonly SCN4A mutation enhances Na inactivation 16 The mutation slows the activation rate of L-type Ca current to 30% of NL Reduced RYR1-mediated Ca release from SER Reduced calcium current density Impaired E-C coupling ? role of K and ? inexcitability Ca homeostasis change reduces ATP-dependent K channel current and leads to abnormal depolarization (Tricarico D et al 1999) 17 Serum K < 3.0mEq/L Serum CK level elevated EKG changes – U waves, flattening of T waves Provocative testing - Intravenous glucose load/ insulin Electrophysiology ◦ Sensory and motor NCS normal between attacks ◦ During attacks – small CMAP. Reduced insertional activity, fibs and positive sharp waves ◦ No myotonia on EMG ◦ Short/ long exercise test 18 Muscle biopsy reserved to atypical patients with normal provocative and gene testing Vacuoles reflect permanent myopathy Vacuoles represent proliferation, degeneration and autophagic destruction of T-tubules & SR Large central vacuoles in hypokalemic PP 19 20 Reducing exposure to known triggers Acute treatment – replacement of K Acetazolamide – prevent attack recurrence and severity ◦ Acetazolamide may ppt weakness in HypoKPP2 Dichlorphenamide – no longer available Triamterene and spironolactone 21 R/O secondary forms Measure K+ during attack Provocative testing for PP: seldom done! o Hypo: gluc/insulin o Hyper: K+ Muscle Bx – vacuoles/dilated T-tubules Electrophysiology o EMG o Short/long exercise tests Genetics 22 More useful in MC Baseline CMAP Exercise 10 sec Record CMAP immediately post exercise, then q 10 sec for 1 min. CMAP in MC and PMC PMC exacerbated by cold No change in CMAP in HypoKPP (Streib. Musc. Nerve. 1982; 5: 719-723) (Fournier. Ann. Neurol. 2004; 56: 650-661) (Fournier Neurology 2009) 23 Record ulnar CMAP Amp baseline Exercise ADM 5 min Check CMAP every 2 min. for 50 min In PP (all types), Amp immed post ex, over next 10-40 min, grad dec amp In MC ↓ Amp immed post ex, rapid return to baseline In PMC sustained ↓ Amp (McManis. Musc. Nerve. 1986; 9: 704-710) (Fournier. Ann. Neurol. 2004; 56: 650-661) 24 Dr.Barohn’s presentation on “Muscle Channelopathies” Anthony A.Amato and James A.Russell; Non dystrophic myotonias and periodic paralysis. Neuromuscular disorders 2008, Mc Graw Hill, Section II Chapter 29; 655-680 Burge JA, Hanna MG. Novel insights into the pathomechanisms of skeletal muscle channelopathies; Curr Neurol Neurosci Rep. 2012 Feb Vol 12:62-69 Hanna, Dipa L Raja Rayan and Michael G. Skeletal Muscle Channelopathies:Non Dystrophic Myotonias and Periodic Paralysis. Current Opinion in Neurology, 2010 Vol. 23: 466-476 neuromuscular.wustl.edu Doreen Fialho and Michael G.Hanna. Periodic paralysis, Handbook of Clinical Neurology, 2007 Vol. 86 (3rd series), p 89-90 25