4._Degenerative_Motor_Neuron_Diseases
advertisement

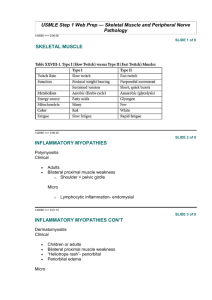
Prepared by: Dr. Sarwer Jamal Bajalan M.B.Ch.B, F.I.B.M.S(Neurology) 2014 Essentials of diagnosis Weakness No sensory loss or sphincter disturbance Progressive course. No identifiable underlying cause other than genetic basis in familial cases. General Considerations Characterized clinically by weakness and variable wasting of affected muscles, without accompanying sensory changes. MND generally commences between 30 and 60 years of age. There is degeneration of: ○ Anterior horn cells in the spinal cord, the motor nucle of the lower cranial nerves, and ○ Corticospinal and corticobulbar pathways. The disorder is usually sporadic, but familial cases may occur Cigarette smoking may be one risk factor. Classification Five varieties have been distinguished on clinical grounds. A. Progressive Bulbar Palsy B. Pseudobulbar Palsy C. Progressive Spinal Muscular Atrophy D. Primary Lateral Sclerosis E. Amyotrophic Lateral Sclerosis (ALS) A mixed UMN and LMN deficit is found in the limbs. ALS (Prof Stephen Hawking( Infantile spinal muscular atropy SMA1 Symptoms and Signs In Bulbar type ○ Dysphagia , dyspea , and dysarthria. ○ Drooping of the palate; a depressed gag reflex; pooling of saliva in the pharynx; a weak cough; and a wasted, fasciculating tongue. In pseudobulbar palsy, the tongue is contracted and spastic and cannot be moved rapidly from side to side. Weakness , stiffness, wasting & fasciculations (UMN &/or LMN) No objective changes on sensory examination. The sphincters are generally spared. Cognitive changes or pseudobulbar affect may be present. The disorder is progressive, and ALS is usually fatal within 3–5 years. Death usually results from pulmonary infections. Patients with bulbar involvement generally have the poorest prognosis, Primary lateral sclerosis often have a longer survival. Ix EMG: chronic partial denervation, with abnormal spontaneous activity in the resting muscle and a reduction in the number of motor units under voluntary control. Confident Diagnosis of ALS needs changes to be found in at least: o Three spinal regions (cervical, thoracic, lumbosacral) or o Two spinal regions and the bulbar musculature. The serum creatine kinase may be slightly elevated. Genitics: D Dx Monoclonal gammopathies. Multifocal nmotor neuropathies with conduction block. Hodgkin disease. Infective anterior horn cell diseases (polio virus or West Nile virus infection). Treatment Riluzole, 50 mg orally twice daily, which reduces the presynaptic release of glutamate, may slow progression of amyotrophic lateral sclerosis. There is otherwise no specific treatment Symptomatic and supportive measures may include: ○ Anticholinergic drugs (such as trihexyphenidyl, amitriptyline, or atropine). ○ Portable suction machine . ○ Physical therapy to prevent contractures. ○ Spasticity may be helped by baclofen or diazepam. ○ A semiliquid diet or nasogastric tube feeding MYOPATHIC DISORDERS DISORDERS Muscular Dystrophies Essentials of diagnosis ○ Muscle weakness, often in a characteristic distribution. ○ Age at onset and inheritance pattern depend on the specific dystrophy General Considerations These inherited myopathic disorders are characterized by progressive muscle weakness and wasting. They are subdivided by mode of inheritance, age at onset, and clinical features. In Duchenne Muscular Dystrophy: ○ ○ ○ ○ ○ ○ ○ ○ Pseudohypertrophy of muscles frequently occurs at some stage; Intellectual retardation is common; and there may be Skeletal deformities, Muscle contractures, and Cardiac involvement. X-linked recessive Age at Onset 1-5 year Pelvic, then shoulder girdle; later, limb and respiratory muscles are affected. ○ Rapid progression (death within about 15 years after onset). Ix The serum creatine kinase(CK) level is increased. EMG & histopathology may help confirm that weakness is myopathic rather than neurogenic. A genetic defect on the short arm of the X-chromosome has been identified in Duchenne dystrophy. The affected gene codes for the protein dystrophin, which is markedly reduced or absent from the muscle of patients with the disease. Dystrophin levels are generally normal in the Becker variety, but the protein is qualitatively altered. Facioscapulohumeral Gowers’ sign showing a patient using arms to climb up thelegs in attempting to get up from the floor Lordotic posturing Managemennt There is no specific treatment for the muscular dystrophies, but it is important to encourage patients to lead as normal lives as possible. Prednisone (0.75 mg/kg orally daily) improves muscle strength and function in boys with Duchenne dystrophy, but side effects need to be monitored. Prolonged bed rest must be avoided, as inactivity often leads to worsening of the underlying muscle disease. Physical therapy and orthopedic procedures may help counteract deformities or contractures. Myotonic Dystrophy(DM) A slowly progressive, dominantly inherited disorder, Usually manifests itself in the 3rd or 4th decade but occasionally appears early in childhood. Myotonic dystrophy type 1 results from an expanded CTG repeat in a protein kinase gene on chromosome 19. In Myotonic dystrophy type 2, the defect is a CCTG repeat expansion in the gene for zinc-finger protein-9 on chromosome 3. EMG o Myotonic discharges o Myopathic changes DM1 Clinical Features od MD1 Muscle stiffness is evidenced by the marked delay of relaxation after muscle contraction. This can often be demonstrated clinically by delayed relaxation of the hand after sustained grip or by percussion of the belly of a muscle. In addition, there is weakness and wasting of the facial, sternocleidomastoid, and distal limb muscles. Associated clinical features include: ○ ○ ○ ○ ○ ○ ○ Ptosis Cataracts , Frontal baldness, Testicular atrophy, Diabetes mellitus, Cardiac abnormalities, and Intellectual changes. Treatment When myotonia is disabling, treatment with a sodium channel blocker—such as: ○ Phenytoin ○ Procainamide ○ Mexiletine Neither the weakness nor the course of the disorder is influenced by treatment. INFLAMMATORY MYOPATHIES: Inclusion Body Myositis This disorder, of unknown cause, begins insidiously, usually after middle age, with progressive proximal weakness of first the lower and then the upper extremities, and affecting facial and pharyngeal muscles. Weakness often begins in the quadriceps femoris in the lower limbs and the forearm flexors in the upper limbs. Distal weakness is usually mild( small mm of the hands). Serum CK levels may be normal or increased. The diagnosis is confirmed by muscle biopsy. Corticosteroid and immunosuppressive therapy is usually ineffective, but IVIG therapy is occasionally of mild benefit. Polymyositis & Dermatomyositis ----- Myopathies Associated with Other Disorders Muscle weakness may be caused by a range of metabolic, endocrine, toxic or inflammatory disorders . Disorders affecting the muscles’ structural integrity can be distinguished by EMG from those caused by metabolic derangement. In metabolic disorders, weakness is often acute and generalised, while a proximal myopathy predominantly affecting the pelvic girdle is a feature of some endocrine disorders. This may develop without other manifestations of hormonal disturbance. A wide variety of drugs and toxins may cause myopathy Causes of acquired proximal myopathy PERIODIC PARALYSIS SYNDROMES (channelopathies) Periodic paralysis may have a familial (dominant inheritance) basis. Episodes of flaccid weakness or paralysis, sometimes in association with abnormalities of the plasma potassium level. Strength is normal between attacks. Hypokalemic periodic paralysis characterized by attacks that tend to occur on awakening, after exercise, or after a heavy meal and may last for several days. Patients should avoid excessive exertion. Hyperkalemic periodic paralysis Normokalemic periodic paralysis Chanalopathies Treatment of Hypokalemic PP A low carbohydrate and low-salt diet may help prevent attacks, as may acetazolamide, 250–750 mg An ongoing attack may be aborted by potassium chloride given orally or by intravenous drip, provided the ECG can be monitored and kidney function is satisfactory. In young Asian men, it is commonly associated with hyperthyroidism. Treatment of the endocrine disorder prevents recurrences.