Glomerular Diseases: Nephrotic & Nephritic Syndromes
advertisement

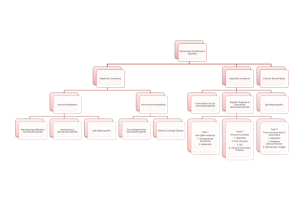
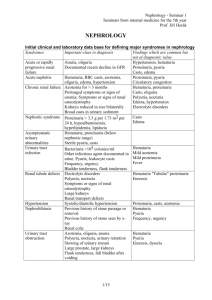
GLOMERULAR DISEASES: NEPHROTIC AND NEPHRITIC SYNDROME Prof Dr. Gülçin Kantarcı Yeditepe University, Medical Faculty Nephrology Department Aims & objectives State the definition of nephritic syndrome. Identify clinical signs of nephritic syndrome. Explain the pathophysiology of nephritic syndrome. State the definition of nephrotic syndrome. Identify clinical signs of nephrotic syndrome. Explain the pathophysiology of nephrotic syndrome. REFERENCE 1. Current Medical Diagnosis and Treatment, Maxine A. Papadakis, Stephen J. McPhee, Eds. Michael W. Rabow, Associate Ed. http://Chapter 22. Glomerular Diseases 2. Bates' Guide to Physical Examination & History Taking 11th edition, Bickleys LS, Szilagyi PG; Lippincott Williams and Wilkins¸ 3. Kumar and Clark's Clinical Medicine, 8th edition; Kumar & Clark, Elsevier 4. Andreoli and Carpenter's Cecil Essentials of Medicine 8th edition, Andreoli and Carpenter, Elsevier PART 11: RENAL AND GENITOURINARY DISEASES 123: Glomerular Disorders and Nephrotic Syndromes 5. http://www.uptodate.com -Differential diagnosis and evaluation of glomerular disease - Overview of heavy proteinuria and the nephrotic syndrome Glomerulus the glomerular capillaries can be injured in a variety of ways, producing many different lesions and several unique changes to urinalysis. There are many forms of glomerular disease with pathogenesis variably linked to the presence of genetic mutations, infection, toxin exposure, autoimmunity, atherosclerosis, hypertension, emboli, thrombosis, or diabetes mellitus. Even after careful study, however, the cause often remains unknown, and the lesion is called idiopathic or primary. NEPHROTIC SYNDROME The nephrotic syndrome is caused by renal diseases that increase the permeability across the glomerular filtration barrier Two issues are important in the pathogenesis of nephrotic syndrome: the mechanisms of glomerular injury and proteinuria. Components of nephrotic syndrome Proteinuria 3.5g/24 hr/1.73m2 Hypoalbum. ( serum alb<3.5 g/d) Hypercholesterolemia (>200mg/dl) Peripheral edema ± anasarca Tendencies of glomerular diseases to manifest Nephrotic Features Minimal change glomerulopathy Membranous GN Diabetic GS Amyloidosis Focal segmental GS Fibrilary GN Common causes of NS Primary glomerular disorders Minimal Change Disease FSGS Membranous GN Orthostatik or postural proteinuria Idiopatic MPGN IgA nephropathy Proliferatif GN Common causes of NS Secondary glomerular disorders Hereditary-familial: DM, Alport’s Syndrome, Sickle cell disease Autoimmun: SLE, Goodpasture’s syndrome, Wegener’s granulomatosis, PAN, RA Infectious: postinfectious glomerulonephritis, HIV FSGS Drug-induced: NSAIDs, Heroin,gold, mercury, Neoplastic: Hodgkin’s D., Lymphomas, leukemia, MM. miscellaneous: amyloidosis, preeclampsia-eclampsia, renovascular HT, intertitial nephritis, fever, exercise Diabetic nephropathy (1/3 cause in patient on dialysis) Nodular Sclerosis in DM Screening for Diabetic nephropathy When BP each visit Urinary alb: Type 2: annually, beginning at diagnosis Type 1: annually, 5 years post diagnosis Normal Range <130/80 <30 mg/day <20 mic/min <30 mic/mg creatinine ADA 2004 Proteinuria podocyte changes to capillary endothelial cells, the glomerular basement membrane (GBM), or podocytes, which normally filter serum protein selectively by size and charge. The mechanism of damage to these structures is unknown in primary glomerular disease The result is urinary loss of macromolecular proteins, primarily albumin but also opsonins, Ig’s, erythropoietin, transferrin, hormone-binding proteins, and antithrombin III in conditions that cause nonselective proteinuria. Pathophysiology of proteinuria 1. 2. Glomerular retention and leakage of protein molecules Damage of glomeruler size and charge selectivity Size:(molecular radius) <17 Å readly pass the glomerular filter, >44 Å can not pass) Albumin 36 Å Charge: glomerular capillary wall fix negative charge Normally 1500mg/24 hr Protein filtered, most is reabsorbed <150 mg of protein excreted each day in the urine Types of proteinuria 'Glomerular' proteinuria (more than about 1.5 g protein/24 h, mostly albumin) 'Tubular' proteinuria (never excretion of more than 1.5 g/24 h ) 'Overflow' proteinuria: immunoglobulin light chains in the urine Benign proteinuria 'Jogger's nephritis' , orthostatic proteinuria Acute Complications of Nephrotic Syn. peripheral edema, ascites, and effusions increased risk for infection (especially cellulitis and, in 2 to 6%, spontaneous bacterial peritonitis); anemia; abnormal thyroid function; Thromboembolism (especially renal vein thrombosis and pulmonary embolism in up to 5% of children and 40% of adults). Thromboembolism may develop not only because of urinary loss of antithrombin III but also because of increased hepatic synthesis of clotting factors, platelet abnormalities, and hyperviscosity from hypovolemia. Hypercholesterolaemia is present in 90 per cent of patients with a urinary protein excretion of over 3 g/24 h Hyperlipidemia in the NS is the result of both increased synthesis and decreased catabolism of lipoproteins. Chronic complications of NS malnutrition in children, coronary artery disease in adults, chronic renal failure, and bone disease. Malnutrition may mimic kwashiorkor, including brittle hair and nails, alopecia, and stunted growth. Coronary artery disease develops because NS causes hyperlipidemia, hypertension, and hypercoagulability. Chronic complications of NS Bone disease develops because of vitamin D deficiency and corticosteroid use. hypothyroidism from loss of thyroid-binding globulin proximal tubular dysfunction causing glucosuria, aminoaciduria, K depletion, phosphaturia, and renal tubular acidosis Symptoms and Signs anorexia, malaise, and frothy urine caused by high concentrations of protein. Edema may cause dyspnea (pleural effusion or laryngeal edema), chest discomfort (pericardial effusion), arthralgia (hydrarthrosis), abdominal pain (ascites or, in children, mesenteric edema). Edema may obscure signs of muscle wasting and cause parallel white lines in fingernail beds (Muehrcke's lines). Diagnosis suspected in patients with edema and proteinuria on urinalysis and confirmed by 24-h measurement of urinary protein. The cause may be suggested by history (eg, cancer); when the cause is unclear, serologic testing and renal biopsy are indicated. Besides proteinuria, urinalysis may demonstrate RBCs and casts (hyaline, granular, fatty, waxy, RBC, or epithelial cell). Lipiduria, the presence of free lipid or lipid within tubular cells (oval fat bodies), within casts (fatty casts), or as free globules Oval Fat Body Evaluation for secondary causes serum glucose or glycosylated hemoglobin (HbA1c), antinuclear antibodies, hepatitis B and C serologic tests, Renal Bx In adults, a renal biopsy is indicated to diagnose the underlying cause of idiopathic NS. Idiopathic NS in children is most likely minimal change disease and is usually presumed without biopsy unless the patient fails to improve on a trial of corticosteroids. NEPHRITIC SYNDROME Defined by hematuria and RBC casts on microscopic examination of urinary sediment. Components of ?Nephritic Syndrome Often one or more elements of mild to moderate proteinuria, edema, hypertension, elevated serum creatinine, and oliguria are also present. It has both primary and secondary causes. Diagnosis is based on history, physical examination, and sometimes renal biopsy. Treatment and prognosis vary by cause. •Asymptomatic Hematuria •Recurrent Gross Hematuria • • • • • Red blood cell/ HP mic >3 cell Asymptomatic Hematuria prevalence 5-10 % Tea or Cola colored urine Proteinuria< 1 g/24 hr Serum Cr< 1.5 mg/dl Tendencies of glomerular diseases to manifest Nephritic Features Proliferative GN(SLE) Acute diffuse proliferative GN (Post Step. GN) Crescentic GN(SLE, Wegener, Ig A) Mesangioproliferative GN(Ig A) Asymptomatic glomerular hematuria • • • • No pathologic abnormality (30%) Thin basement membrane (26%) Ig A nephropathy (28%) Alport syndrome ( rare ? Is it really rare in Turkey ?) Alport's Syndrome Hereditary nephritis is a genetically heterogenous disorder characterized by hematuria, impaired renal function, sensorineural deafness, and ocular abnormalities. Cause is a gene mutation affecting type IV collagen. Symptoms and signs are those of nephritic syndrome with sensorineural deafness and, less commonly, those of ophthalmologic diseases. Diagnosis is by family history and urinalysis. Alport's Syndrome Because of X-linked transmission, women usually are asymptomatic and have little functional impairment. Most men eventually develop renal symptoms and signs similar to those of acute nephritic syndrome and progress to renal insufficiency between ages 20 and 30. Alport's Syndrome Diagnosis is suggested by personal and family history and by findings of microscopic hematuria on urinalysis or recurrent episodes of gross hematuria, particularly if abnormalities of hearing or vision are present. IgA nephropathy It is the most common form of GN worldwide. It occurs at all ages, with a peak onset in the teens and 20s; affects men 2 to 6 times more frequently than women; and is more common in whites and Asians than in blacks. Prevalence estimates are 5% in the US, 10 to 20% in southern Europe and Australia, and 30 to 40% in Asia. IgA nephropathy Deposition of IgA immune complexes in glomeruli, manifesting as slowly progressive hematuria, proteinuria, and, often, renal insufficiency. Diagnosis is based on urinalysis and renal biopsy. Prognosis is generally good ?. Treatment options include ACE inhibitors, corticosteroids, and ω-3 polyunsaturated fatty acids. PATHOGENESIS Cause is unknown, pathogenetic mechanisms, increased IgA1 production, defective IgA1 glycosylation causing increased binding to mesangial cells, decreased IgA1 clearance, a defective mucosal immune system, overproduction of cytokines stimulating mesangial cell proliferation. Familial clustering has also been observed, suggesting genetic factors at least in some cases. Symptoms and Signs persistent or recurrent macroscopic hematuria (90% of involved children) or asymptomatic microscopic hematuria with mild proteinuria Diagnosis urinalysis and renal biopsy. Urinalysis demonstrates microscopic hematuria, usually with dysmorphic RBCs and RBC casts. Mild proteinuria (< 1 g/day) is typical and may occur without hematuria; nephrotic syndrome develops in ≤ 20%. Ig A mesangial deposition granular deposition of IgA and C3 on immunofluorescent staining in an expanded mesangium with foci of segmental proliferative or necrotizing lesions. Postinfectious Glomerulonephritis most common cause of glomerular disease in children between 5 and 15 yr; it is rare in children < 2 yr and in adults > 40 yr. occurs after infection, usually with a nephritogenic strain of group A β-hemolytic streptococcus. Diagnosis is suggested by history and urinalysis and confirmed by low complement. Symptoms and signs asymptomatic hematuria (in about 50%) and mild proteinuria full-blown nephritis with microscopic or gross hematuria (cola-colored, brown, smoky, or frankly bloody), proteinuria, oliguria, edema, hypertension, and renal insufficiency. Severe, late disease is a relatively uncommon cause of nephrotic syndrome. Flank pain may be attributable to stretching of the renal capsule. Renal failure that causes fluid overload with heart failure and urgent or malignant hypertension and requires dialysis affects 1 to 2% of patients and may present as a pulmonary-renal syndrome with hematuria and hemoptysis Rapidly Progressive Glomerulonephritis (RPGN) Crescentic Glomerulonephritis RPGN causes microscopic glomerular crescent formation with progression to renal failure within weeks to months. Diagnosis is based on history, urinalysis, serologic tests, and renal biopsy. Treatment is with corticosteroids, with or without cyclophosphamide, and sometimes plasmapheresis. Classification of RPGN Based on Immunofluore scence Microscopy Anti-glomerular basement membrane (GBM) antibody disease (type 1 RPGN) The combination of GN and alveolar hemorrhage in the presence of anti-GBM antibodies is called Goodpasture's syndrome autoimmune GN and accounts for 10% of RPGN cases. It may arise when respiratory exposures (eg, cigarette smoke, viral URI) expose alveolar capillary collagen, triggering formation of anticollagen antibodies. The anticollagen antibodies cross-react with GBM, fixing complement and triggering a cell-mediated inflammatory response in the kidneys and lungs. Immunofluorescent staining of renal biopsy tissue demonstrates linear IgG deposits. Immune complex RPGN (type 2 RPGN) complicates numerous infectious and connective tissue disorders and also occurs with other primary glomerulopathies. Immunofluorescent staining demonstrates nonspecific granular immune deposits. The condition accounts for 40% of RPGN cases. Pathogenesis is usually unknown. Pauci-immune RPGN (type 3 RPGN) distinguished by the absence of immune complex or complement deposition on immunofluorescent staining. It constitutes 50% of all RPGN cases. Almost all patients have elevated antineutrophil cytoplasmic antibodies (ANCAs) and systemic vasculitis crescent formation in a patient with ANCA–associated small vessel vasculitis Crescentic Glomerulonephritis Thin Basement Membrane Disease (Benign Familial Hematuria) Thin basement membrane disease is diffuse thinning of the glomerular basement membrane from a width of 300 to 400 nm in normal subjects to 150 to 225 nm. Renal disease in patients with hematuria undergoing renal biyopsy Prot<1 Cr<1.5 Prot 1-3 Cr 1.5-3 Cr >3 No abnormality 30% 2% 1% 0% Thin BM nephropathy 26% 4% 3% 0% Ig A Nephropathy 28% 24% 14% 8% GN without crescent 9% 26% 37% 23% GN with crescent 2% 24% 21% 44% Other renal disease 5% 20% 24% 25% Total 100% 100% 100% 100% N=43 N=123 N=179 N=255 Renal biopsy: Indications • • • The cause cannot be determined or adequatly predicted by less invasive diagnostic procedures The sign and symptoms suggest parenchymal disease that can be diagnosis by pathologic evaluation The differential diagnosis includes diseases that have different treatment, different prognosis, or both Renal biopsy: Contraindications • • • • • • • • • • • Solitary kidney Uncooperative patient Bleeding disorders Uncontrolled severe HT Severe anemia or dehidratation Cystic kidney Hydronephrosis Multiple renal arterial aneurysms Acute pyelonephritis or perinephric abscess Renal neoplasm ESRD