Learning Objectives")
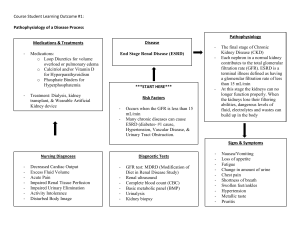
I’m Still Not Feeling Well (PBL 2) PBL Learning Objectives Describe the anatomical and physiological determinants of glomerular filtration rate and renal blood flow Glomerular filtration rate (GFR) and renal blood flow (RBF) are intimately related. Anatomical Determinants of Renal Blood Flow Blood Supply Each kidney receives about 10% of the total blood volume per minute (receive 1200 mL/min) via the large renal arteries, which are lateral branches of the abdominal aorta, just inferior to the origin of the superior mesenteric artery (L1-L2). The right renal artery is longer and passes posterior to the IVC. The renal artery divides into five separate segmental arteries, which then move into the different segments of the kidneys and do not anastomose with each other. This results in distinct vascular segmentation of the kidney, with each being surgically resectable. Several renal veins drain each kidney and unite in a variable fashion to form the right and left renal veins, which lie anterior to the right and left renal arteries. To note: Efferent arterioles arising from cortical nephrons divide into two peritubular capillaries that surround the proximal (PCT) and distal convoluted tubules (DCT), forming a rich meshwork of microvessels (which function to remove water and solutes from renal tubule). This drains into the interlobular veins. Efferent arterioles arising from juxtamedullary nephrons (nephrons closest to medulla) give rise to vasa recta, which descends with the loop of Henle into the renal medulla and return to area of glomerulus before draining into interlobular or arcuate veins - Each vasa recta carrier blood at a very slow rate, which is crucial in countercurrent exchange that prevents the washout of concentration gradients established in the urine. Gradient maintenance is important in the kidney’s ability to concentrate urine. - On the descending part of vasa recta, NaCl and urea are reabsorbed into the blood and water is secreted. On the ascending portion of vasa recta, NaCl and urea are secreted into the interstitium and water is reabsorbed The Renal Corpuscle and the Glomerulus The nephron is the basic unit of kidney structure and function, each containing a: 1. Renal corpuscle, which filters fluid from the blood. The renal corpuscle consists of a compact tuft of interconnected capillary loops, the glomerulus, surrounded by a hollow capsule (Bowman’s Capsule). 2. A long, renal tube that converts filtered fluid into urine The glomerulus is an anastomosing network of capillaries composed of fenestrated endothelial cells. The capillaries have a high hydrostatic pressure (~60 mmHg) that causes rapid fluid filtration. The renal corpuscle has a urinary pole (proximal tube attaches to the glomerulus) and a vascular pole (arterioles attach and macula densa is located). The urinary pole is where there is an abrupt change (but continuous) from flat epithelial cells of Bowman’s capsule to cuboidal epithelial cells of PCT. The vascular pole is where the glomerulus is connected to afferent and efferent arterioles. Afferent arterioles are larger than efferent to help maintain filtration pressure within glomerulus. Arterioles are associated with juxtaglomerular, which is composed of: - Macula Densa consists of closely spaced epithelial cells of the distal tubule. These cells sense sodium concentration in the tubular lumen. - Juxtaglomerular Cells are modified smooth muscle cells found mainly in the afferent arteriole, which sense pressure changes in the arterioles and secrete renin. - Lacis Cells (Mesangial Cells) which have an uncertain role but may produce erythropoietin. The Glomerular Filtration Barrier The filtration barrier in the renal corpuscle, through which all filtered substances must pass has: 1. The endothelial cells of the capillaries are perforated by many large fenestrae. Although these fenestrations are fairly large, the endothelial cells are well endowed with fixed negative charges that hinder the passage of plasma proteins. 2. The basement membrane/basal lamina, which is consists of a meshwork of collagen and proteoglycans fibrillae that have large spaces through which large amounts of water and small solutes can filter. This basement membrane effectively prevents the filtration of plasma proteins due to the strong negative electrical charges associated with proteoglycans. 3. The podocytes (epithelial cells) that rest of the basement membrane and face Bowman’s space. They are attached to the basement membrane via foot processes, which have filtration slits (25-60 nm diameter) present between them. 4. This final layer is not continuous, but sends long secondary members that encircle the outer surface of the capillaries. The secondary members are separated by gaps called slit pores through which the glomerular filtrate moves. These epithelial cells, which also have negative charges, provide additional restriction to the filtration of plasma proteins Physiological Determinants of GFR- Glomerular Filtration and GFR The glomerular filtration process results in the formation of an ultrafiltrate of plasma A key function of the glomerulus is to act as a filtration barrier that permits the passage of water and other solutes and restricts the movement of certain molecules. Selectivity of filtration is dependent on molecule size, as well as charge Glomerular filtration rate (GFR) describes the flow rate of filtered fluid through the kidney. It is equal to the sum of all the filtration rates of all the functioning nephrons, and as such, is an index of kidney function In normal adults, the GFR averages 90-140 mL/min for males, and 80-125 mL/min for females. Thus, approximately 180L/day of plasma is filtered at the glomerulus. The GFR is determined by: - The balance of hydrostatic and colloid osmotic forces acting across the capillary (net filtration pressure) - The glomerular capillary coefficient (KP), which is the product of the permeability and filtering surface area of the capillaries. The normal KP is about 12.5 mL/min/mmHg of filtration pressure. GFR= KP x Net Filtration Pressure Ultrafiltration occurs because Starling forces (i.e. hydrostatic and oncotic pressures) drive fluid from the lumen of the glomerular capillaries, across the filtration barrier, and into Bowman’s space: - Hydrostatic pressure inside the glomerular capillaries, which promotes filtration of fluid into Bowman’s space - Hydrostatic pressure in Bowman’s capsule outside the capillaries, which opposes filtration - Colloid osmotic pressure of the glomerular capillary plasma proteins, which opposes filtration - Colloid osmotic pressure of the proteins in Bowman’s capsule The glomerular ultrafiltrate is essentially protein free and the oncotic pressure in Bowman’s space is essentially zero In this way, the GFR can be altered by changing KP or any of the Starling forces Increased KP raises GFR and vice versa- Some diseases lower KP by reducing the number of functional glomerular capillaries, lowering the surface area for filtration Increasing Bowman’s Capsule Hydrostatic Pressure Decreases GFR and vice versa- In certain pathological states associated with obstruction of the urinary tract, Bowman’s capsule pressure can increase markedly Increasing glomerular capillary colloid oncotic pressure, decreases GFR- Increasing arterial plasma colloid oncotic pressure or increasing the filtration fraction (fraction of plasma filtered by glomerular capillaries) raises the glomerular colloid osmotic pressure decreasing GFR, as there is an increased tendency to keep fluid in the capillaries Increased glomerular capillary hydrostatic pressure increases GFR- Changes in glomerular hydrostatic pressure serve as the primary means for physiologic regulation of GFR. - The GFR is governed by three interplaying factors in regards to the afferent and efferent arterioles of the nephron. - The aortic pressure perfusing the kidney (kidney perfusion)- Increased arterial pressure tends to raise glomerular hydrostatic pressure, and therefore, increase GFR. However this effect is buffered by autoregulatory mechanisms that maintain a relatively constant glomerular pressure as blood pressure fluctuates. - Increased resistance of afferent arterioles reduced glomerular hydrostatic pressure and decreases GFR, as it determines the degree of renal arterial pressure transmitted to the glomerulus. - Constriction of the efferent arterioles increases the resistance to outflow from the glomerular capillaries. This raises the glomerular hydrostatic pressure, but also reduces renal blood flow, causing the filtration fraction and glomerular colloid osmotic pressure increase as efferent arteriolar resistance. Thus, efferent arteriolar constriction has a biphasic effect on GFR. At moderate levels of constriction, there is a slight increase in GFR, but with severe constriction, there is a decrease in GFR - A fall in resistance to outflow from effect glomerular capillaries decreases the glomerular capillary hydrostatic pressure, and thus decreases GFR Renal Blood Flow and Oxygen Consumption The kidneys normally consume oxygen at twice the rate of the brain but have almost seven times the blood flow of the brain. Thus, the oxygen delivered to the kidneys far exceeds their metabolic needs, and the arterial-venous extraction of oxygen is relatively low compared with that of most other tissues. A large fraction of the oxygen consumed by the kidneys is related to the high rate of active sodium reabsorption by the renal tubules – i.e. if renal blood flow and GFR are reduced and less sodium is filtered, less sodium is reabsorbed and less oxygen is consumed The outer part of the kidney, the renal cortex, receives most of the kidney’s blood flow (98-99%) Flow to the renal medulla is supplied by a specialized portion of the peritubular capillary system called the vasa recta Determinants of Renal Blood Flow Renal blood flow is determined by the pressure gradient across the renal vasculature: Renal artery pressure is about equal to systemic arterial pressure, and renal vein pressure averages about 3 to 4 mm Hg under most conditions Most of the renal vascular resistance resides in three major segments: interlobular arteries, afferent arterioles, and efferent arterioles. Resistance of these vessels is controlled by the sympathetic nervous system, various hormones, and local internal renal control mechanisms. An increase in the resistance of any of the vascular segments of the kidneys tends to reduce the renal blood flow, whereas a decrease in vascular resistance increases renal blood flow if renal artery and renal vein pressures remain constant.. Autoregulation of Renal Blood Flow and GFR Feedback mechanisms intrinsic to the kidneys normally keep the renal blood flow and GFR relatively constant, despite marked changes in arterial blood pressure (autoregulation) Autoregulatory mechanisms aim to preserve changes in arterial blood pressure, which are the essential driving forces for glomerular filtration This ensures precise control of renal excretion of water and solutes Two mechanisms are responsible for autoregulation of RBF and GFR: one mechanism that responds to changes in arterial pressure (myogenic mechanism), and another that responds to changes in the NaCl concentration of tubular fluid (tubuloglomerular feedback) The Role of the Tubuloglomerular Feedback in Autoregulation of GFR To perform the function of autoregulation, the kidneys have a feedback mechanism that links changes in sodium chloride concentration at the macula densa with the control of renal arteriolar resistance This feedback helps ensure a relatively constant delivery of sodium chloride to the distal tubule and helps prevent spurious fluctuations in renal excretion that would otherwise occur The tubuloglomerular feedback mechanism has two components that act together to control GFR: - An afferent arteriolar feedback mechanism - An efferent arteriolar feedback mechanism These feedback mechanisms depend on special anatomical arrangements of the juxtaglomerular complex, which consists of macula densa cells in the initial portion of the distal tubule and juxtaglomerular cells in the walls of the afferent and efferent arterioles The macula densa cells sense changes in volume delivery to the distal tubule by way of signals that are not completely understood. Studies suggest that decreased GFR slows the flow rate in the loop of Henle, causing increased reabsorption of sodium and chloride ions in the ascending loop of Henle, thereby reducing the concentration of sodium chloride at the macula densa cells This decrease in sodium chloride concentration initiates a signal from the macula densa that has two effects: - It decreases resistance to blood flow in the afferent arterioles, which raises glomerular hydrostatic pressure and helps return GFR toward normal - It increases renin release from the juxtaglomerular cells of the afferent and efferent arterioles. Renin released from these cells increases the formation of angiotensin I, which is converted to angiotensin II. Finally, the angiotensin II constricts the efferent arterioles, thereby increasing glomerular hydrostatic pressure and returning GFR toward normal Myogenic Autoregulation of Renal Blood Blow and GFR Another mechanism that contributes to the maintenance of a relatively constant renal blood flow and GFR is the ability of individual blood vessels to resist stretching during increased arterial pressure, a phenomenon referred to as the myogenic mechanism. Individual blood vessels (especially small arterioles) throughout the body respond to increased wall tension or wall stretch by contraction of the vascular smooth muscle Stretch of the vascular wall allows increased movement of calcium ions from the extracellular fluid into the cells, causing them to contract This contraction prevents over distension of the vessel and at the same time, by raising vascular resistance, helps prevent excessive increases in renal blood flow and GFR when arterial pressure increases. Sympathetic Nervous System Activation Decreases GFR Essentially all the blood vessels of the kidneys, including the afferent and the efferent arterioles, are richly innervated by sympathetic nerve fibres Strong activation of the renal sympathetic nerves can constrict the renal arterioles and decrease renal blood flow and GFR Moderate or mild sympathetic stimulation has little influence on renal blood flow and GFR i.e. reflex activation of the SNS resulting from moderate decreases in pressure at the carotid sinus baroreceptors has little influence on renal blood flow or GFR. The renal sympathetic nerves seem to be most important in reducing GFR during severe, acute disturbances lasting for a few minutes to a few hours, such as those elicited by the defence reaction, brain ischemia, or severe hemorrhage In the healthy resting person, sympathetic tone appears to have little influence on renal blood flow. Angiotensin II Constricts Efferent Arterioles Angiotensin II is produced systemically and within the kidney It constricts the afferent and (mainly) efferent arterioles and decreases RBF and GFR Angiotensin-converting enzyme (ACE) degrades and thereby inactivates bradykinin and converts angiotensin I, an inactive hormone, to angiotensin II. Thus, ACE increases angiotensin II levels and decreases bradykinin levels Drugs called ACE inhibitors, which reduce systemic blood pressure in patients with hypertension, reduce angiotensin II levels and elevate bradykinin levels. These effects lower systemic vascular resistance, reduce blood pressure, and decrease renal vascular resistance. ACE inhibitors therefore increase RBF and GFR Fall in renal perfusion pressure Activation of renin-angiotensin system AT2 preferentially locally increases resistance at efferent arteriole = vasoconstriction Preservation of GFR in hypotension Neurohumoral Influences Sympathetic stimulation can also cause vasoconstriction due to stimulation of alpha-1-adrenergic receptors by noradrenaline. Reduced renal perfusion pressure in patients is most often due to depletion of effective circulating volume (e.g. hypovolemia or dehydration), causes systemic hypoperfusion, which leads to increased release of vasoconstrictors AT2 and Noradrenaline. As mentioned, AT2 increases efferent arteriole resistance; whereas noradrenalin increases tone in both arterioles. The net effect is renal vasoconstriction in order to shunt blood to the critical coronary and cerebral circulations whilst maintaining GFR (sympathetic response). Hormone Mechanism Final Effect Noradrenaline Angiotensin II Endothelin Prostaglandins (E and I) Nitric Oxide Bradykinin Atrial Natriuretic Peptide Glucocorticoids Dopamine Histamine Vasoconstriction of afferent and efferent arterioles In low concentrations, constricts efferent arteriole, in high concentrations constricts both Secreted by endothelial cells of renal vessels causing vasoconstriction of afferent and efferent arterioles. It contributes to haemostasis when a blood vessel is severed, when the endothelium is damaged. Produced locally in kidneys during conditions such as haemmorrhage. They inhibit vasoconstrictive role of sympathetic stimulation and angiotensin II RBF/GFR RBF/GFR Vasodilates afferent and efferent arterioles Stimulates release of prostaglandins and NO As heart is stretched due to increased cardiac load, atrial cells secrete ANP which dilates afferent and efferent arterioles RBF/GFR RBF/GFR RBF/GFR Produced by proximal tubule and increases RBF and inhibits rennin secretion Decreases resistance of afferent and efferent arterioles RBF/GFR RBF/GFR RBF/GFR RBF/GFR RBF/GFR Describe the function of the renal tubules, i.e. Reabsorption, secretion and urine concentration Overview Out of the 180 L of glomerular filtrate former per day, about 1.5 L (<1%) is excreted as urine. Selective reabsorption and absorption of solutes and water along the renal tubule facilitate this low urine output. The tubules precisely control volume, osmolality, composition and pH of intracellular and extracellular fluid compartments. Transport mechanisms across cell membrane: - Passive Transport Simple Diffusion along electrochemical gradients (both in charge and concentration) and can follow either a transcellular or intercellular route Facilitated diffusion, where the movement of one solute along an electrochemical gradient facilitates the movement of another against its electrochemical gradient. - Active Transport This involves movement of an ion against electrochemical gradient, which requires energy expenditure. The two central functions of renal tubules are: - Tubular reabsorption- active transport of solutes e.g. glucose, amino acids, electrolytes and vitamins from glomerular filtrate, through the tubular lumen and into peritubular capillaries - Tubular Secretion- transport of solutes from peritubular capillaries into tubular lumen Proximal Tubular Transport This region of the nephron has a high energy consumption and: 1) Is the “workhorse” of sodium and water reabsorption (65-75% of the filtered load) 2) Reabsorbs almost all filtered glucose, phosphate, amino acids and other organic solutes by linking their transport to glucose PCT cells are characterised by brush borders on their luminal surfaces, composed of thousands of villi which increase the absorptive surface area significantly. Absorbed substances are transferred to the peritubular capillaries from the basolateral membrane of the cell. Sodium Ion Reabsorption- This is done through a combination of active and passive transport, though mainly active via a basolateral Na+-K+-ATPase pump, which sets up an electrochemical gradient for passive solute diffusion, osmosis and secondary active transport (cotransport) with Na +. It pumps 3Na+ out of cell and 2 K+ into cell The combination of low [Na+] and cell-interior negative potential results in favorable electrochemical gradient for sodium entry into the cell across the apical membrane - In the early PCT, Na+, glucose and other organic solutes, enter the cell with Na + and leave the cell across the basolateral membrane by passive transport mechanism. Na + is reabsorbed by cotransport with H+ or organic solutes (glucose, amino acids, phosphate and lactate). - In the late PCT, Na+ is absorbed via chloride-driven sodium transport mechanism across both transcellular and paracellular pathways. In the paracellular pathway, the high concentration of Cl- in the late PCT creates a concentration gradient that facilitates diffusion of Cl - with Na+ through the PCT cell. In the transcellular pathway, Na+ reabsorption occurs via parallel operation of Na +H+ and one or more Cl- anion (formate) antiporters. The transporter results in secretion of H+ ion into the tubular lumen in exchange for a Na+. Much of the secreted H+ then combines with bicarbonate, leading to the reabsorption of 90% of filtered bicarbonate Osmotic water transport occurs due to the high permeability of apical and basolateral membranes to water due to presence of aquaporins (transmembrane water channels). The removal of solutes from the lumen initially lowers tubular fluid osmolality, thereby creating an osmotic gradient, which promotes an equivalent degree of water reabsorption. Water reabsorption also occurs between the cells across leaky tight junctions on the proximal tubule. The sodium concentration and osmolality of the fluid leaving the proximal tubule is the same as that of plasma Disorders in proximal tubule transport are quite rare, and are associated with: - Glycosuria - Amino aciduria - Type 2 “proximal” renal tubular acidosis (impaired acid excretion) Loop of Henle This region of the nephron reabsorbs approximately 20-25% of the filtered Na+ (and thus Cl-) and is relatively impermeable to water movement (lacking aquaporins). As a result, solutes are reabsorbed in greater quantity than water, diluting the tubular fluid and increasing the solute concentration of the medulla, contributing to the concentrating function of the collecting duct. Thin Descending Limb- Water absorption occurs passively (due to hypertonic interstitial fluid) exclusively in this part of the Loop of Henle. This is accompanied by diffusion of Na + ions from interstitial fluid into tubular lumen. Thin Ascending Limb – Limited passive reabsorption of Na + and Cl-. Thick Ascending Limb – impermeable to H2O but reabsorbs 20-25% of filtered Na +, Cl- and other cations. - Na+, K+, 2Cl- cotransporter mediated active transport of sodium. Site of action of Loop Diuretics (e.g. furosemide) which inhibit the Na+, K+, 2Cl- cotransporter Paracellular passive reabsorption of Na+, K+, Ca2+ and Mg2+ between cells across tight junction - Chloride exits the cell through a basolateral channel Distal Tubule Distal tubule reabsorbs 5-8% of filtered sodium chloride and is relatively impermeable to water, leading to a fall in [Cl-]. This limits sodium chloride reabsorption in the Loop of Henle and distal tube. - Activity of Na-K-2Cl and Na-Cal cotransporters is primarily determined by luminal chloride concentration. Thus, a reduction in chloride concentration will reduce rate of sodium chloride entry into the cell. This is because the binding of chloride to this transporter induces a conformational change that is required for solute movement into the cell - Sodium chloride concentration in the peritubular interstitium is similar to plasma. Hence, falling concentration within the lumen creates a favourable concentration for backflux of sodium and chloride into lumen across tight junctions. Reabsorption ceases when the rate of sodium entry into the cell equals the rate of backflux The major site of urine dilution is the cortical diluting segment, where solutes are absorbed more than water. The electrochemical gradient is driven by the Na+-K+ ATPase, with a luminal Na+-Cl- cotransporter. Thiazide diuretics block the Na+-Cl- cotransporter, which may lead to excessive dilution of body fluids Calcium Transport- Distal tubules has major sites at which urinary calcium excretion is actively regulated under the influence of parathyroid hormone and vitamin D Collecting Duct There are two distinct cell types: - Principal - Intercalated (type A and type B) This region of the nephron is responsible for concentrating the urine (i.e. water reabsorption), as well as acid and potassium excretion. Vasopressin stimulates the insertion of water channels (aquaporin’s) into the luminal and basal membranes, and hence water is absorbed across the epithelium layer. Principal Cells- In the cortical collecting tubule and the cells in the inner medullar collecting duct play an important role in sodium and water reabsorption and potassium secretion Solute Transport in the Principal Cell The electrochemical gradient is driven by the Na+-K+ ATPase pump, where the principal cell has an overall negative charge, excess potassium and relative deficiency of sodium. Sodium is absorbed into the cell via a luminal transporter, hence transferring the negative charge to the lumen. Potassium then leaves via the apical and luminal channels (luminal excretion is favoured by the negative charge). Expression of both channels is under the control of aldosterone, a hormone which increases DNA transcription and also ANP, which decreases Na+ reabsorption by reducing the number of open Na+ channels. Intercalated cells- In the cortex and the cells in the outer medulla are primarily involved in the regulation of acid-base balance Transport in the Type A Intercalated Cell These cells are used to excrete H+ ions into the urine via: - Active H+ pump - K+-H+ exchanger (plays only a minor role) On the basolateral membrane, the Cl--HCO3- exchanger is used to reabsorb bicarbonate back into the bloodstream. The net effect is excretion of H+ ions, thereby acidifying the urine. Transport in the Type B Intercalated Cell This is simply the “mirror image” of the type A intercalated cell. These cells are used to excrete bicarbonate and reabsorb hydrogen, with the net effect being reabsorption of H + ions from the urine. Describe the renal actions and regulation of the renin-angiotensin system, prostaglandins and atrial natriuretic peptide Overview The renin-angiotensin-aldosterone axis promotes Na+ retention through the actions of both angiotensin II and aldosterone. It plays an important role in regulating blood volume and systemic vascular resistance, which together influence cardiac output and arterial pressure It is regulated by the mechanisms that stimulate renin release Angiotensinogen, and Angiotensin I and II Angiotensinogen is synthesised by the liver and released into the systemic circulation. The liver contains only small stores of Angiotensinogen Another protein, renin, is produced and stored in distinctive granules by the granular cells of the juxtaglomerular apparatus. Decreases in effective circulating volume stimulate these cells to release renin. This protease cleaves Angiotensinogen, thereby releasing angiotensin I (ANG I) Angiotensin-converting enzyme (ACE) rapidly removes two C-terminal amino acids from the physiologically inactive ANG I to form ANG II. ACE is present on the luminal surface of vascular endothelium throughout the body, and is abundantly present in endothelium-rich lungs. ACE in the kidney. particularly in the endothelial cells of the afferent and efferent arterioles, can produce enough ANG II to exert local vascular effects Thus, the kidney receives ANG II from two sources: - Systemic ANG II comes from the general circulation and originates largely from the pulmonary region - Local ANG II forms from the renal conversion of systemic ANG II Renin Release The principal factor controlling plasma ANG II levels is renin release from JGA granular cells A decrease in effective circulating volume manifests itself to the JGA, and thus stimulates renin release in three ways: Decreased systemic blood pressure: - A low effective circulating volume, sensed by baroreceptors located in the central arterial circulation, signals medullary control centres to increase sympathetic outflow to the JGA, thus increasing renin release - Renal denervation or β-adrenergic blocking drugs (e.g. propanolol) inhibit renin release Decreased NaCl concentration at macula densa (NaCl sensor): - Independent of renal nerve activity or renal perfusion pressure, decreased effective circulating volume decreases GFR and thus reduces luminal [NaCl] at the macula densa, thereby increasing renin release Decreased renal perfusion pressure (renal baroreceptor): - Stretch receptors in the granular cells of the afferent arterioles sense the decreased distension associated with low effective circulating volume - This decreased stretch lowers [Ca2+]I, thereby increasing renin release and initiating a cascade that tends to increase blood pressure - Conversely, increased distension (higher extracellular volume) inhibits renin release Some additional factors also modulate renin release: - Prostaglandins E2 and I2 and endothelin all activate renin release - Agents that blunt renin release ANG II (which represents a short feedback loop), AVP, thromboxane A2, high plasma levels of K+ and NO Actions of Angiotensin II Stimulation of Aldosterone release from glomerulosa cells in the adrenal cortex: - Aldosterone tends to promote Na+ and water retention and lowers plasma K+ concentration. It acts on the principal cells in the distal tubule and the collecting duct, up-regulating and activating the basolateral Na+-K+ pumps, hence increasing Na+ reabsorption (and water) into the blood and secreting K+ into the urine. - In animal models, unopposed aldosterone infusion causes increased glomerulosclerosis and severe proteinuria through a non-epithelial profibrotic effect on the kidney. The aldosterone component of RAAS can be targeted with mineralocorticoid receptor blockers, offering benefits additive to the therapy of ACE inhibitors. Vasoconstriction of renal and other systemic vessels: - ANG II increases Na+ reabsorption by altering renal haemodynamics, probably in two ways - First, at high concentrations, ANG II constricts the efferent more than the afferent arterioles, thus increasing filtration fraction and reducing the hydrostatic pressure in the downstream peritubular capillaries. The increased filtration fraction also increases the protein concentration in downstream blood and hence raises the colloid osmotic pressure of the peritubular capillaries. This favours the uptake of reabsorbate from the peritubular interstitium into peritubular capillaries, and hence enhance the reabsorption of Na+ and fluid by the proximal tubule - Second, ANG II decreases medullary blood flow through vasa recta. This decreases medullary washout of NaCl and urea, a process that raises [urea] in the medullary interstitium and enhances Na+ reabsorption along the thin ascending limb of Henle’s loop ANG II works by binding to AT1 receptors at the apical and basolateral membranes of proximal tubule cells, and stimulating apical NHE3s ANG II also stimulates Na-H exchange in the thick ascending limb (TAL) and stimulates apical Na+ channels in the initial collecting tubule These effects promote sodium reabsorption Enhanced tubuloglomerular feedback: - ANG II raises the sensitivity and lowers the set point of the tubuloglomerular feedback mechanism, so that an increase in Na + and fluid delivery to the macula densa elicits a much more pronounced fall in the GFR Enhanced Na-H exchange: - ANG II promotes Na+ reabsorption in the proximal tubule, thick ascending limb (TAL), and initial collecting tubule Renal hypertrophy: - ANG II induces hypertrophy of renal tubule cells Stimulated thirst and AVP (ADH/vasopressin) release: - ANG II acts on the hypothalamus, where it increases the sensation of thirst and stimulates secretion of ADH from the posterior pituitary, both of which increase total body free water - This ANG II effect represents an intersection between the systems for regulating effective circulating volume and osmolality ADH acts by binding to a V2 receptor at the basolateral membrane of target cells, which increases cAMP In the TAL, ADH stimulates the apical NKCC2 and K + channels In principal cells, ADH stimulates Na+ transport by increasing the number of open Na+ channels in the apical membrane ACE Inhibitors These drugs are used to control hypertension by decreasing the overall levels of angiotensin II. It has several tissue and renal effects: - Reverse hypertrophy and vascular remodelling in hypertensive disease - Reverse rarefaction in vascular disease - Decrease protein excretion in many renal diseases - Decrease the production of aldosterone (acutely) - Limit aldosterone’s capacity for potassium excretion (in exchange for sodium in the distal tubule) The drug almost invariably decreases renal plasma flow and the GFR. Angiotensin Receptor Antagonists These drugs decrease the tissue efficacy of angiotensin II. The overall effects include: Decrease cardiac remodelling Decrease GFR Decrease renal protein excretion In general, drugs that act upon the renin-angiotensin-aldosterone system have a beneficial effect on blood pressure, providing a 5-10 mmHg further reduction, whilst also reducing proteinuria by 30-55%. However, there is a 0-10% risk of severe hyperkalaemia. Atrial Natriuretic Peptide (ANP) Of the main methods that correct a low circulating volume, ANP is the only one that does so by decreasing its activity. It is as an important counter-regulatory system As its name implies, ANP promotes natriuresis (i.e. Na+ excretion) Atrial myocytes synthesise and store ANP and release ANP in response to stretch (a low-pressure volume sensor). Thus, reduced circulating volume inhibits ANP release and reduces Na + excretion ANP has many synergistic effects on renal haemodynamics and on transport by renal tubules that promote renal Na+ and water excretion Although ANP directly inhibits Na+ transport in the inner medullary collecting duct, its major actions are haemodynamic- increased GFR, increased cortical and medullary blood flow, and decreased release of renin and AVP. Increased flow to medullary interstitium decreases osmolality and ultimately reduces passive Na+ reabsorption in the thin ascending limb Thus, a decrease in effective circulating volume leads to a fall in ANP release and a net decrease in Na + and water excretion ANP binds to ANP-A-receptors on collecting duct cells and acts via cGMP to inactivate apical sodium channels, so reducing sodium reabsorption It also inhibits aldosterone release and renin production and increases GFR by dilating afferent arterioles Prostaglandins Both COX-1 and COX-2 are present in the kidney in constitutive and inducible forms The prostaglandins that are most important in the kidney are PGE2 and PGI2 - PGE2 decreases sodium reabsorption at the thick ascending limb of the loop of Henle - PGI2 prostacyclin stimulates renin release, which in turns increases aldosterone. Prostacyclin is also a strong vasodilator that maintains glomerular filtration rate (GFR) and renal blood flow in patients with decreased actual or effective circulating volume, however the vasodilatory role of PGI2 is not operative and has little importance in renal haemodynamics. As a result of inhibiting PGE2, one may experience: - Sodium retention, manifested as weight gain - Peripheral oedema - Congestive heart failure (rarely) - Blood pressure may also increase because of sodium retention This occurs most often in patients with hypertension who are being treated with antihypertensive medications. In addition, inhibition of PGI2 can cause hyperkalaemia or, in patients at risk for adverse renal effects, acute renal failure NSAIDs can be grouped into four categories: 1. Selective COX-1 inhibitors, such as aspirin 2. Non-selective COX inhibitors 3. Relatively selective COX-2 inhibitors 4. Highly selective COX-2 inhibitors Describe the role of the kidney in acid-base and potassium balance, and the consequences of hyperkalaemia Acid-Base Balance If pH deviates too far from the normal range, cells become poisoned by their own toxic waste and die. Imbalanced pH interrupts all cellular activities and functions and can lead to the progression of most degenerative diseases, such as cardiovascular disease, cancer, diabetes and weight gain. There is normally a tight control over the H+ concentration, where pH is normally maintained between 7.35 – 7.45. Metabolic acid production (e.g. sulphuric and phosphoric) and H+ intake must be balanced by acid excretion. There are three central mechanisms that are involved in the removal of H+ and elimination from the body There are two classes of acids that are physiologically important, an sources of H+ are listed below: Carbonic acids are gnerated from metabolism of fat and carbohydrates. They are potentially produced from accumulation of CO2 in the body and are primarily eliminated by alveolar ventilation Non-carbonic acids are generated from protein metabolism, and their elimination is a two-step process. It involves the initial combination with extracellular bicarbonate and intracellular buffers to minimise change in free hydrogen concentration. Then there is the subsequent excretion of acid by the kidney Defence against Shift in pH The first line of defence is the acid-base buffer systems. The secondary line of defence includes: - Renal excretion of H+ - Respiratory excretion of CO2 The respiratory centre responds to H+ increases in the following way: - If pH is low (acidic), breathing can increase, thus removing H+ - If pH is high (basic), breathing can decrease, thus adding H+ 1. Physiological Buffers Buffers are primarily weak acids, which are able to take up or release H+ so that changes in free H+ concentration are minimised. They are located in extracellular fluids, intracellular fluids and bone. Extracellular Buffers include: - Bicarbonate/carbon dioxide buffer system - Inorganic phosphates (plasma phosphate concentration of 1 mmol/L versus 24 mmol/L of HCO3-) - Plasma proteins HCO3- is the most important buffer in extracellular fluid: - It is relatively high in concentration - Ability to vary pCO2 via changes in alveolar ventilation - Concentration is measurable - Bicarbonate that is used in the buffering process must be regenerated to maintain normal acidbase balance. For example, acidaemia stimulates an increase in ventilation, which blunts the pH change, but does not regenerate HCO3-. - As these buffer systems are in equilibrium, altering the bicarbonate system will change body pH, which resets the ratio of acid to base in the other buffers - The lungs alter the bicarbonate system by altering PCO2 and the kidneys by altering the HCO3Intracellular and Bone Buffers - These include proteins, organic and inorganic phosphate and haemoglobin. - The bones are an important site for the buffering of acid and base loads - An acid load, for example, is associated with the uptake of some of the excess H + ions by bone, resulting in the release of buffer compounds, such as CaCO3 and CaHPO4, into the extracellular fluid. This is demonstrated by decreased osteoblastic and increased osteoclastic function, resulting in bone loss. - The main intracellular buffers are sodium phosphate (Na 2HPO4/NaH2PO4) and proteins proteins can act as acids or bases because they contain both acidic and basic amino acid side chains 2. Renal Excretion of Hydrogen (Reabsorption of filtered HCO 3-) Virtually all filtered HCO3- must be reabsorbed before dietary H+ load can be excreted 1. Intracellular H2O breaks down into H+ and OH- ions 2. OH- combines with CO2 to form HCO3- via a reaction catalysed by CA 3. In the proximal tubule, H+ ions are secreted into the lumen by the Na-H exchanger and HCO3- ions are returned to systemic circulation by a Na+-3HCO3- cotransporter. 4. In collecting tubules, the processes are mediated by the active H+-ATPase pump and the Cl—HCO3exchanger. 5. Secreted H+ ions combined with the filtered HCO 3- to form H2CO3, and then CO2 and H2O, which are passively reabsorbed. The dissociation of carbonic acid is facilitated when luminal carbonic anhydrase is present. The net effect is HCO3- reabsorption, although the HCO3- returned are not the same as those filtered. The collecting tubules also have H+-K+-ATPase pumps in the luminal membrane, which are involved in both acid secretion and K+ reabsorption. Ammonia Handling and Acid-Base Balance Tubular cells, principally those in the proximal tubule, metabolise glutamine to produce ammonia and, ultimately, glucose and bicarbonate The bicarbonate enters the blood and the NH4+ ions (which effectively carry a H + ion) are excreted in the urine NH3 enters the filtrate from the tubular cells by simple diffusion or by the Na +/H+ exchanger, which can also transport NH4+ once in the lumen, NH3 is protonated to NH4+, which cannot diffuse out of the tubules In the thick ascending limb of the loop of Henle, NH4+ can be transported out of the lumen in place of K+ on the NaK2Cl co-transporter Also, as the tip of the loop of Henle is alkaline, NH4+ in the filtrate dissociates to form NH3 and this diffuses into the interstitium Secreted protons may also combine with ammonia either way, this ultimately results with the addition of acid to the tubular fluid and the extraction of acid from the ECF Acid-Base Disorders Acidosis is a process that tends to lower extracellular fluid pH. - Metabolic – low pH and low HCO3- concentration - Respiratory – low pH and high pCO2 Alkalosis is a process that tends to raise the extracellular fluid pH. - Metabolic – high pH and high HCO3- concentration - Respiratory – high pH and low pCO2 Potassium Balance Potassium is the major intracellular cation, with an intracellular concentration of around 150 mmol/L, compared with around 4 mmol/L in ECF. To maintain potassium balance, the kidney excretes only 5-15% of filtered potassium. Potassium, like sodium, is freely filtered in the glomerulus, but is handled quite differently in the tubule. Almost all the filtered potassium is reabsorbed before the filtrate reaches the collecting tubules, where any potassium that is to be excreted is then secreted into the collecting duct In order to maintain appropriate intracellular potassium concentration, all cells use a pump-leak mechanism. This consists of the Na+/K+ ATPase pump (major driving force behind potassium movement) which actively transports potassium into the cell, balanced by various channels which allow potassium to leak out of the cell. Intracellular potassium can be controlled by changing the activity of the pump or by altering the number of permeability of the potassium channels Potassium regulation takes place in two steps - Initial uptake of some of the ingested potassium into cells, limiting rise in plasma potassium concentration after ingestion - Subsequent excretion of excess potassium in the urine – within 6-8 hours Potassium uptake by cells- In normal people, 3 factors are significant in promoting potassium uptake by cells - Small elevation in plasma potassium concentration - Insulin - Adrenalin (epinephrine) Basal levels of insulin and adrenalin maintain activity of Na-K-ATPase pump and increases in either hormone STIMULATES pump activity Adrenalin released during stress response can also significantly drive potassium into luminal cells and lower plasma [K+] Urinary potassium excretion - Urinary potassium is NOT from glomerular filtration - Almost all filtered potassium is reabsorbed passively in the proximal tubule and thick ascending limb of the loop of Henle. The rate of potassium excretion being primarily determined by potassium secretion from the cell into the lumen in the principal cells in the cortical collecting tubule and outer medullary collecting tubule Potassium secretion occurs through selective potassium channels in the apical membrane - This selective process is enhanced by reabsorption of sodium through selective sodium channels in the apical membrane - Removal of sodium from lumen creates a lumen-negative electrical potential that promotes potassium SECRETION through the apical potassium channels AND CHLORIDE REABSORPTION between cells across the tight junction Aldosterone plays central role in regulation of potassium excretion - A small rise in plasma K+ is enough to increase the adrenal release of aldosterone - Aldosterone then enters the potassium-secreting cell in the distal nephron and combines with its cytosolic receptor – this complex then migrates to nucleus where it initiates synthesis of aldosterone induced proteins - Aldosterone enhances potassium secretion by affecting each of the steps involved in this process Distal delivery of water and sodium - At a constant aldosterone and plasma [K+], increasing sodium delivery (eg loop diuretic) will tend to enhance distal sodium re-absorption and THEREFORE potassium EXCRETION - Converse applies – decreased distal delivery of Na+ will diminish potassium secretion = predisposing to hyperkalemia Consequences of Hyperkalemia The normal plasma potassium concentration is 3.5-5.0 mmol/L. As potassium is the main determinant of the resting membrane potential of excitable cells, disturbances of plasma potassium can cause cardiac dysrhythmias or arrest. Hyperkalaemia usually represents reduced urinary potassium excretion or, less commonly, acute release from cells or a failure to enter cells. Hyperkalaemia does not persist unless there is impaired renal excretion Causes Shifts Out of Cells: - During metabolic acidosis, H+ ions enter cells to be buffered and K + ions leave the cells to maintain electroneutrality - Insulin deficiency in diabetic ketoacidosis allows the net movement of potassium out of the cells - Rhabdomyolosis or tissue destruction, or lysis such as that caused by chemotherapy, can cause massive potassium loss from cells Failure of Renal Secretion: - In renal failure, potassium accumulates because of the reduced number of nephrons capable of potassium excretion Other Causes of Hyperkalaemia: - Trimethoprim and pentamidine therapy can both can block K+ secretion in the collecting tubule - Hypoaldosteronism- inadequate renin release, inadequate aldosterone release, or tubular resistance to aldosterone. It is usually caused by potassium-sparing diuretics hyporeninemic Treatment The patient should be placed on a cardiac monitor. If there are any ECG changes, urgent treatment is essential Initially, calcium, given as calcium gluconate or calcium chloride, will antagonise the effects of potassium on the cardiac action potential, but this is short-lived In the intermediate term, potassium can be shifted into cells by administering insulin, combined with glucose, to prevent hypoglycaemia. β2 agonists can also be used for this purpose. Administration of sodium bicarbonate produces a temporary alkalosis which also promotes the intracellular movement of potassium In the longer term, excess potassium must be removed from the body. Diuretics, such as furosemide, combined with hydration encourage renal excretion. If renal function is severely impaired, dialysis or haemofiltration will remove potassium. Cation exchange resins such as sodium polystyrene sulfonate can be given orally or rectally and bind potassium in the guy, exchanging it for sodium Describe the mechanisms involved in fluid and electrolyte balance, thirst and salt appetite The normal sodium intake in a Western diet is 150-200 mmol/L and 95 mmol/day recommended for a healthy heart The normal potassium intake is 60-80 mmol/day or 200-400 mmol/day on a vegetarian diet For the average 70kg male, the daily fluid intake should be around 2.5 L. Disturbances to the normal electrolyte balance can result in the following symptoms: Thirst Muscle weakness Neurological impairment (Hallucination, Decreased skin turgor Delirium, Seizures) Haemodynamic effects Cardiac disturbances – ECG changes High risk of falls Volume Homeostasis- Osmoreceptor (ADH Feedback System) When osmolarity (plasma sodium concentration) increases above normal because of water deficit (hypernatremia), for example, a feedback system kicks in. The hypothalamic osmoreceptors which release ADH are stimulated by hyperosmolality and inhibited by hypoosmolality. Large hypothalamic cells around the third ventricle send their axons directly to the posterior pituitary, where the axon terminals release oxytocin and vasopressin (ADH) into the bloodstream. ADH secretion is influenced by several factors (note that anything that stimulates ADH secretion also stimulates thirst): 1. By special receptors in the hypothalamus that are sensitive to increasing plasma osmolarity (when the plasma gets too concentrated). These stimulate ADH secretion. 2. By stretch receptors in the atria of the heart, which are activated by a larger than normal volume of blood returning to the heart from the veins. These inhibit ADH secretion, because the body wants to rid itself of the excess fluid volume. 3. By stretch receptors in the aorta and carotid arteries, which are stimulated when blood pressure falls. These stimulate ADH secretion, because the body wants to maintain enough volume to generate the blood pressure necessary to deliver blood to the tissues. ADH (vasopressin) is transported to all parts of the body by the circulation, but the major site of action is the kidneys. In the collecting duct, it bind to the V2 receptors on the principal cells of the collecting duct, causing an increase in water permeability by inducing water channels (aquaporin’s) stored in intracellular vesicles to fuse with the luminal membrane. The effect on water permeability is graded- the higher the ADH concentration, the more receptor sites occupied and the greater the water permeability. The increased water permeability in the distal nephron segments causes increased water reabsorption and excretion of a small volume of concentrated urine ADH also increases the urea permeability of the collecting duct principal cells. Furthermore, ADH has a rapid action and rapid turnover (10-20 minutes). Renal Sodium Handling Tubular reabsorption of Na AII Sympathetic tone Unloading of baroreceptors Aldosterone Renin release ECFV Contraction NORMAL ECFV ECFV expansion ANF Dopamine Renal perfusion pressure Renal interstitial hydrostatic pressure Renal prostaglandins Tubular reabsorption of Na Sodium is the major extracellular cation and its concentration is tightly controlled Sodium and chloride ions are freely filtered in the glomerulus, so the concentration of these ions in the filtrate is similar to that in blood (135-145mmol/L) The kidney reabsorbs a huge amount of salt in the proximal tubules and the loop of Henle, and the little that is left is reabsorbed in a precisely regulated manner by the distal tubules and collecting ducts to maintain accurate salt balance. About 5% of salt intake is also lost in faeces and sweat The basolateral membranes of the tubular cells contain Na+/K+ ATPases that actively pump sodium into the peritubular plasma. From here, they pass freely into the blood to complete the reabsorption process The continual pumping of sodium out of cells and its subsequent removal by the blood creates a Na + gradient between the tubular filtrate and the cell cytoplasm. This gradient allows Na + from the filtrate to enter the cells passively at their apical membrane, provided that suitable channels or transporters are present Role of Thirst in Controlling Extracellular Fluid Osmolarity and Sodium Concentration The kidneys minimize fluid loss during water deficits through the osmoreceptor-ADH feedback system. Adequate fluid intake, regulated by the thirst mechanism, is necessary to counterbalance whatever fluid loss does occur through sweating and breathing and through the gastrointestinal tract. Central Nervous System Centres for Thirst The same area along the anteroventral wall of the third ventricle that promotes ADH release also stimulates thirst. Located anterolaterally in the preoptic nucleus is another small area that, when stimulated electrically, causes immediate drinking that continues as long as the stimulation lasts. All these areas together are called the thirst centre. The neurons of the thirst centre respond to injections of hypertonic salt solutions by stimulating drinking behaviour. These cells almost certainly function as osmoreceptors to activate the thirst mechanism, in the same way that the osmoreceptors stimulate ADH release. Increased osmolarity of the cerebrospinal fluid in the third ventricle has essentially the same effect to promote drinking. Stimuli for Thirst One of the most important is increased extracellular fluid osmolarity, which causes intracellular dehydration in the thirst centres, thereby stimulating the sensation of thirst. This helps to dilute extracellular fluids and returns osmolarity toward normal. Decreases in extracellular fluid volume and arterial pressure also stimulate thirst by a pathway that is independent of the one stimulated by increased plasma osmolarity. Thus, blood volume loss by hemorrhage stimulates thirst even though there might be no change in plasma osmolarity. This probably occurs because of neutral input from cardiopulmonary and systemic arterial baroreceptors in the circulation. A third important stimulus for thirst is angiotensin II.. Because angiotensin II is also stimulated by factors associated with hypovolemia and low blood pressure, its effect on thirst helps to restore blood volume and blood pressure toward normal, along with causing the kidneys to decrease fluid excretion. Dryness of the mouth and mucous membranes of the esophagus can elicit the sensation of thirst. Salt-Appetite Mechanism for Controlling Extracellular Fluid Sodium Concentration and Volume Maintenance of normal extracellular fluid volume and sodium concentration requires a balance between sodium excretion and sodium intake. Nowadays, most people eat far more sodium than is necessary for homeostasis, and there is evidence that our usual high sodium intake may contribute to cardiovascular disorders such as hypertension. There is also a regulatory component to salt appetite in which there is a behavioural drive to obtain salt when there is sodium deficiency in the body. In general, the two primary stimuli that are believed to increase salt appetite are - Decreased extracellular fluid sodium concentration - Decreased blood volume or blood pressure, associated with circulatory insufficiency Describe the volume and composition of body fluid compartments and principles of rehydration therapy Overview The total body fluid is distributed mainly between the extracellular fluid and the intracellular fluid - The extracellular fluid is divided into the interstitial fluid and the blood plasma - There is also another small compartment of fluid that is referred to as transcellular fluid. This compartment includes fluid in the synovial, peritoneal, pericardial, and intraocular spaces, as well as the cerebrospinal fluid In the average 70-kilogram adult human, the total body water is about 60 % of the body weight (~42 litres). This percentage can change, depending on age, gender, and degree of obesity As a person grows older, the percentage of total body weight that is fluid gradually decreases. This is due in part to the fact that aging is usually associated with an increased percentage of the body weight being fat, which decreases the percentage of water in the body Intracellular Fluid Compartment The intracellular fluid constitutes about 40% of the total body weight in an “average” person. The fluid of each cell contains its individual mixture of different constituents, but the concentrations of these substances are similar from one cell to another, and it is for this reason, the intracellular fluid of all the different cells together is considered to be one large fluid compartment. Extracellular Fluid Compartment All the fluids outside the cells are collectively called the extracellular fluid. Together these fluids account for about 20% of the body weight, or about 14 liters in a normal 70 kg adult The two largest compartments of the extracellular fluid are the interstitial fluid, which makes up more than three fourths of the extracellular fluid, and the plasma, which makes up almost one fourth of the extracellular fluid, or about 3 litres. Constituents of Extracellular and Intracellular Fluids Comparisons of the composition of the extracellular fluid, including the plasma and interstitial fluid, and the intracellular fluid are shown in this graph below: Rehydration Therapy The aim is to re-establish body fluid homeostasis from a current state of imbalance e.g. in dehydration (hypovolemia) or an oedematous state (hypervolemia) In oedematous patients, hypertonic solutions are sometimes infused into the bloodstream to draw out excess water out of the extracellular space and move it into the bloodstream for kidney excretion. Water is drawn out of the extracellular space due to the process of osmosis, it wants to move down a concentration gradient to create an equilibrium with the hypertonic solution In dehydrated patients, hypotonic solutions may be used to rehydrate their tissues. They are already hypertonic due to the increased concentration of solutes within their body fluid, and hence a hypotonic (i.e. dilute) solution can be used to reach an iso-osmotic state again There are also other situations involving rehydration which have other special considerations: - Haemorrhage treat with infusion of whole blood or a combination of red cells and plasma substitute (e.g. Haemacel) - Plasma loss (i.e. burns) treat with human plasma or plasma substitute - Loss of water and electrolytes (i.e. vomiting, diarrhoea or excessive renal losses) treat with oral replacement of water and sodium salts. Also, IV fluids may be required – rapid infusion (1000ml/hr) is necessary if there is hypotension and/or evidence of impaired organ perfusion. Fluid replacement must always be monitored, particularly in patients suffering from severe hypovolaemia, as overly aggressive rehydration may result in circulatory overload i.e. blood vessels don’t vasodilate quick enough. Also, in any rehydration therapy, solute and electrolyte composition of the replacing fluid should be selected appropriately to maintain osmotic equilibrium between body fluid compartments. Describe the categories of renal failure (prerenal, renal and postrenal) and the immediate consequences of acute renal failure Overview Acute renal failure Prerenal Intrinsic Tubular Acute tubular necrosis Postrenal Glomerular Vascular Glomerulonephritis Vasculitis Ischaemic Toxic Interstitial nephritis Renal failure generally refers to any impairment in the glomerular filtration rate. A fall in GFR with intrinsic renal disease usually reflects disease progression with reduction in number of functioning nephrons Acute renal failure is characterised by: - Decreased renal perfusion - Ischemic, toxic or obstructive insult to the renal tubule - Inflammation and oedema of tubule The causes of acute renal failure can be divided into three main categories- prerenal, postrenal and intra-renal - Acute renal failure resulting from decreased blood supply to the kidneys; this condition is often referred to as prerenal acute renal failure to reflect the fact that the abnormality occurs in a system before the kidneys. This can be a consequence of heart failure with reduced cardiac output and low blood pressure or conditions associated with diminished blood volume and low blood pressure, such as severe haemorrhage - Intra-renal (renal) acute renal failure resulting from abnormalities within the kidney itself, including those that affect the blood vessels, glomeruli, or tubules - Post-renal acute renal failure, resulting from obstruction of the urinary collecting system anywhere from the calyces to the outflow from the bladder. The most common causes of obstruction of the urinary tract outside the kidney are kidney stones, caused by precipitation of calcium, urate, or cystine. Prerenal Acute Renal Failure Decreased renal blood flow is usually accompanied by decreased GFR and decreased urine output of water and solutes. - Oliguria, which refers to diminished urine output below the level of intake of water and solutes, can occur. This causes accumulation of water and solutes in the body fluids. - If renal blood flow is markedly reduced, total cessation of urine output can occur, a condition referred to as anuria. As long as renal blood flow does not fall below 20-25%of normal, acute renal failure can usually be reversed if the cause of the ischemia is corrected before damage to the renal cells has occurred. Unlike some tissues, the kidney can endure a relatively large reduction in blood flow before actual damage to the renal cells occurs The reason for this is that as renal blood flow is reduced, the GFR and the amount of sodium chloride filtered by the glomeruli (as well as the filtration rate of water and other electrolytes) are reduced. This decreases the amount of sodium chloride that must be reabsorbed by the tubules, which uses most of the energy and oxygen consumed by the normal kidney. Therefore, as renal blood flow and GFR fall, the requirement for renal oxygen consumption is also reduced. When blood flow is reduced below this basal requirement, which is usually less than 20 to 25 per cent of the normal renal blood flow, the renal cells start to become hypoxic, and further decreases in renal blood flow, if prolonged, will cause damage or even death of the renal cells, especially the tubular epithelial cell If the cause of prerenal acute renal failure is not corrected and ischemia of the kidney persists longer than a few hours, this type of renal failure can evolve into intra-renal acute renal failure. Intra-renal Acute Renal Failure Includes abnormalities that originate within the kidney and that abruptly diminish urine output This category of acute renal failure can be further divided into - Conditions that injure the glomerular capillaries or other small renal vessels - Conditions that damage the renal tubular epithelium - Conditions that cause damage to the renal interstitium This type of classification refers to the primary site of injury, but because the renal vasculature and tubular system are functionally interdependent, damage to the renal blood vessels can lead to tubular damage, and primary tubular damage can lead to damage of the renal blood vessels. Acute Renal Failure Caused by Glomerulonephritis Acute glomerulonephritis is a type of intra-renal acute renal failure usually caused by an abnormal immune reaction that damages the glomeruli In about 95% of the patients with this disease, damage to the glomeruli occurs 1 to 3 weeks after an infection elsewhere in the body, usually caused by certain types of group A-beta streptococci, i.e. sore throat, tonsillitis, skin infection - It is not the infection itself that damages the kidneys; - Over a few weeks, as antibodies develop against the streptococcal antigen, the antibodies and antigen react with each other to form an insoluble immune complex that becomes entrapped in the glomeruli, especially in the basement membrane portion of the glomeruli - Once the immune complex has deposited in the glomeruli, many of the cells of the glomeruli begin to proliferate, and large numbers of white blood cells become entrapped in the glomeruli - Many of the glomeruli become blocked by this inflammatory reaction, and those that are not blocked usually become excessively permeable, allowing both protein and red blood cells to leak from the blood of the glomerular capillaries into the glomerular filtrate The acute inflammation of the glomeruli usually subsides in about 2 weeks, and in most patients, the kidneys return to almost normal function within the next few weeks to few months Sometimes, however, many of the glomeruli are destroyed beyond repair, and in a small percentage of patients, progressive renal deterioration continues indefinitely, leading to chronic renal failure Tubular Necrosis as a Cause of Acute Renal Failure Tubular necrosis is the destruction of epithelial cells in the tubules. Acute Tubular Necrosis Caused by Severe Renal Ischemia: - If the ischemia is severe enough to seriously impair the delivery of nutrients and oxygen to the renal tubular epithelial cells, and if the insult is prolonged, damage or eventual destruction of the epithelial cells can occur. - When this happens, tubular cells “slough off” and plug many of the nephrons, so that there is no urine output from the blocked nephron. The affected nephrons often fail to excrete urine even when renal blood flow is restored to normal, as long as the tubules remain plugged - The most common causes of ischemic damage to the tubular epithelium are the prerenal causes of acute renal failure associated with circulatory shock Acute Tubular Necrosis Caused by Toxins or Medications: - These substances have specific toxic actions on the renal tubular epithelial cells, causing death of many of them - As a result, the epithelial cells slough away from the basement membrane and plug the tubules - In some instances, the basement membrane also is destroyed - If the basement membrane remains intact, new tubular epithelial cells can grow along the surface of the membrane, so that the tubule repairs itself within 10 to 20 days. Post-renal Acute Renal Failure Multiple abnormalities in the lower urinary tract can block or partially block urine flow and therefore lead to acute renal failure even when the kidneys’ blood supply and other functions are initially normal If the urine output of only one kidney is diminished, no major change in body fluid composition will occur because the contralateral kidney can increase its urine output sufficiently to maintain relatively normal levels of extracellular electrolytes and solutes as well as normal extracellular fluid volume. With this type of renal failure, normal kidney function can be restored if the basic cause of the problem is corrected within a few hours Chronic obstruction of the urinary tract, lasting for several days or weeks, can lead to irreversible kidney damage. Some of the causes of post-renal acute failure include: - Bilateral obstruction of the ureters or renal pelvises caused by large stones or blood clots - Bladder obstruction - Obstruction of the urethra If the urinary tract is obstructed (e.g. by stones or prostate enlargement), hydrostatic pressure in Bowman’s space increases and consequently GFR decreases. A very high rate of urine output also may be accompanied by an increase in hydrostatic pressure in Bowman’s space and a decrease in GFR because an increased pressure head is required to force a large volume flow down tubules and collecting ducts. Physiologic Effects of Acute Renal Failure A major physiologic effect of acute renal failure is retention in the blood and extracellular fluid of water, waste products of metabolism, and electrolytes this can lead to water and salt overload, which in turn can lead to oedema and hypertension. Excessive retention of potassium, however, is often a more serious threat to patients with acute renal failure, because increases in plasma potassium concentration (hyperkalaemia) to more than about twice normal can be fatal. Because the kidneys are also unable to excrete sufficient hydrogen ions, patients with acute renal failure develop metabolic acidosis, which in itself can be lethal or can aggravate the hyperkalaemia. In the most severe cases of acute renal failure, complete anuria occurs. The patient will die in 8 to 14 days unless kidney function is restored or unless an artificial kidney is used to rid the body of the excessive retained water, electrolytes, and waste products of metabolism. Describe the anatomical relationships of the prostate gland and the effect of prostatic enlargement on urinary outflow Structure of the Prostate Gland The prostate (approximately 3 cm long, 4 cm wide, and 2 cm in depth) is the largest accessory gland of the male reproductive system. It is a single, firm, doughnut-shaped retroperitoneal organ that encircles the neck of the bladder and urethra just inferior to bladder. Two-thirds of the prostate is glandular, and the rest is fibromuscular The fibrous capsule of the prostate is dense and neurovascular, incorporating the prostatic plexuses of veins and nerves. All this is surrounded in turn by the visceral layer of the pelvic fascia, forming a fibrous prostatic sheath that is thin anteriorly, continuous anterolaterally with the puboprostatic ligaments, and dense posteriorly where it blends with the rectovesical septum Fluid from prostate plays a role in : - Activating sperm - 1/3 of semen volume - It is milky and slightly acidic which contains citrate (nutrients), enzymes, and PSA (prostate specific agent) - The secretion from the prostatic gland leaves when the smooth muscle contracts during ejaculation The prostate has: - A base closely related to the neck of the bladder. - An apex that is in contact with fascia on the superior aspect of the urethral sphincter and deep perineal muscles. - A muscular anterior surface, featuring mostly transversely oriented muscle fibers forming a vertical, trough-like hemisphincter (rhabdosphincter), which is part of the urethral sphincter, separated from the pubic symphysis by retroperitoneal fat in the retropubic space. - A posterior surface that is related to the ampulla of the rectum. - Inferolateral surfaces that are related to the levator ani. Although not clearly distinct anatomically, the following lobes of the prostate are traditionally described: - The isthmus of the prostate (commissure of prostate; historically, the anterior) lies anterior to the urethra. It is fibromuscular, the muscle fibres representing a superior continuation of the urethral sphincter muscle, and contains little, if any, glandular tissue. - The inferoposterior (posterior) lobe lies posterior to the urethra and inferior to the ejaculatory ducts; it is readily palpable by digital rectal examination. - The right and left (lateral) lobes on either side of the urethra form the major part of the prostate. - The middle (median) lobe lies between the urethra and the ejaculatory ducts and is closely related to the neck of the bladder. Enlargement of the middle lobe is believed to be at least partially responsible for the formation of the uvula that may project into the internal urethral orifice. Benign Prostatic Hyperplasia (BPH) From 40 yrs of age, the prostate increases by 2.4cm3 per year on average Characterised by hyperplasia of prostatic stromal and epithelial cells, resulting in the formation of large, fairly discrete nodules in the periurethral region of prostate. When sufficiently large, the nodules compress and narrow the urethral canal causing partial or complete obstruction of the urethra. This results in: - Compression of urethra with difficulty in urination - Retention of urine in the bladder with subsequent distension and hypertrophy of the bladder, infection of the urine, development of cystitis and renal infections. The middle lobe usually enlarges the most and obstructs the internal urethral orifice. The more the person strains, the more the valve-like prostatic mass occludes the urethra If the urinary tract is obstructed (e.g. by stones or prostate enlargement), hydrostatic pressure in Bowman’s space increases and consequently GFR decreases. A very high rate of urine output also may be accompanied by an increase in hydrostatic pressure in Bowman’s space and a decrease in GFR because an increased pressure head is required to force a large volume flow down tubules and collecting ducts. Symptoms include – hesitancy, poor prolonged flow, sensation of incomplete emptying, and overflow dribbling. Secondary (irritative) symptoms – urinary frequency, nocturia, urgency of micturition and urge incontinence (sudden and strong need to urinate)not specific to BPH. Long-term consequences- Hydronephrosis (swelling of a kidney due to build-up of urine) or acute retention with secondary UTI, azotemia or uremia (urea in bloow) may develop. Secondary changes in the bladder e.g. hypertrophy, trabeculation (dense collagenous tissue) and diverticulum formation. Treatment – medical or surgical therapy e.g. decreasing fluid intake, esp prior to bedtime; moderating intake of alcohol and caffeine-containing products, following timed voiding schedules. Most commonly used and effective medical therapy is α-blockers which decrease prostate smooth muscle tone via inhibition of α1-adrenergic receptors. Urinalysis The ‘normal’ colour for urine is straw-yellow. Abnormalities are : Cloudy – usually due to the presence of white blood cells, bacteria, mucus, RBCs, fat, epithelial cells or phosphates. It is seen in UTIs. Dark – characteristic of liver disorders such as hepatitis or cirrhosis. Red/Pink – can be due to beets or food colouring, or bleeding from UTIs, enlarged prostate, kidney cancer, bladder tumor, tuberculosis, bladder stones, kidney infection, Wilms' tumor (in children), or hypernephroma (tumour resembling adrenal cortex in kidney). Hemolytic anemia and porphyria can also cause urine to take on these colors. It may also occur after trauma to the kidneys or urinary tract. Dark yellow/orange – use of laxatives, excess carotene consumption NSAIDs The relevance of NSAIDs to renal failure is great because they are capable of inducing allergic interstitial nephritis. This is characterized by oedema and cellular infiltrate within the interstitium. This allergic interstitial nephritis has several other unusual features, in that some drugs, particularly fenoprofen, also induce profuse proteinuria which may provoke a full nephrotic syndrome. The renal failure induced may also be irreversible. Their effects on renal blood flow- In any state of renal underperfusion, like mild cardiac decompensation, volume depletion, salt loss, cirrhosis or a nephrotic syndrome, NSAIDs may cause a profound fall in renal bloodflow and in glomerular filtration. The precise mechanism underlying this dangerous effect is not entirely clear, but it appears that vasodilator prostaglandins- principally PGI2 (prostacyclin). This prostaglandin usually helps, in states of renal hypoperfusion, to maintain cortical bloodflow and glomenrular filtration through effects on the intrarenal distribution of blood. NSAIDs act by reducing prostaglandin biosynthesis through inhibition of cyclooxygenase (COX) which exists as two isoforms (COX-1 and COX-2). Drugs that selectively inhibit COX-2 might be expected to produce effects on renal function similar to nonselective NSAIDs which inhibit both COX-1 and COX-2. Class Learning Objectives Kidney response to reduced renal perfusion RAAS, ADH – effects of ACE inhibitors on this system Impact on electrolyte loss Acid-base balance & potassium balance in kidney and hyperkalemia PGI2 role of prostaglandins in Kidneys Mechanisms by which normal BP is maintained (Renal and Cardiac including ANP) Relation of Prostate to Kidney and urinary flow Urinalysis Osmolarity Anatomical and physiological determinants of glomerular filtration rate and renal blood flow. Function of the renal tubules, i.e. reabsorption, secretion and urine concentration. Categories of renal failure (prerenal, renal and postrenal) and the immediate consequences of acute renal failure. The anatomical relationships of the prostate gland and the effect of prostatic enlargement on urinary outflow. Official Learning Objectives Describe the anatomical and physiological determinants of glomerular filtration rate and renal blood flow Describe the function of the renal tubules, i.e. Reabsorption, secretion and urine concentration Describe the renal actions and regulation of the renin-angiotensin system, prostaglandins and atrial natriuretic peptide Describe the role of the kidney in acid-base and potassium balance, and the consequences of hyperkalaemia Describe the mechanisms involved in fluid and electrolyte balance, thirst and salt appetite Describe the volume and composition of body fluid compartments and principles of rehydration therapy Describe the categories of renal failure (prerenal, renal and postrenal) and the immediate consequences of acute renal failure Describe the anatomical relationships of the prostate gland and the effect of prostatic enlargement on urinary outflow. Keywords and Phrases Acidosis, Anion-gap, Acute tubular necrosis, Acute renal failure, Back pain. Dehydration, Uraemia, Diarrhoea, Renal tubular secretion and reabsorption, Osmolality, Specific gravity, Prerenal renal failure, Postural hypotension, Renal blood flow (RBF), Glomerular filtration rate (GFR), Urine concentration, Urinalysis, Prostate, Renal Casts, Nephrotoxicity, Urine microscopy, Oliguria, ACE inhibitor, Angiotensin, Renin, Antidiuretic hormone, Fluid and electrolyte balance, Prostatic hypertrophy, Tissue turgor, JVP Drugs Used Naproxen, Lisinopril